Plain Language in Informed Consent: A Strategic Guide for Clinical Researchers
This article provides a comprehensive guide for researchers and drug development professionals on implementing plain language in informed consent forms (ICFs).
Plain Language in Informed Consent: A Strategic Guide for Clinical Researchers
Abstract
This article provides a comprehensive guide for researchers and drug development professionals on implementing plain language in informed consent forms (ICFs). It covers the ethical and regulatory foundations, outlines a step-by-step methodology for creating clear and accessible documents, offers solutions for common challenges, and reviews evidence validating the impact of these practices on participant comprehension and study integrity. The guidance is aligned with current regulations, including the Revised Common Rule's requirement for a concise key information section, and is designed to improve the ethical conduct and operational efficiency of clinical research.
The Why: Ethical and Regulatory Imperatives for Plain Language Consent
Informed consent serves as a cornerstone of ethical clinical research, embodying two distinct but interrelated components: the informed consent conversation (ICC) and the informed consent document (ICD) [1]. While the completed form provides essential documentation, the underlying process constitutes a comprehensive dialogue between researchers and potential participants. This process begins at the initial solicitation and continues throughout the research relationship, ending only when the participant's information or biospecimens are no longer used in research [2]. The ethical foundation rests on three core principles derived from the Belmont Report: information provision, comprehension assurance, and voluntariness of participation [3]. Effective implementation requires that consent materials be written in plain language appropriate to the subject population, typically at an 8th grade reading level or lower, to facilitate true understanding [4].
Quantitative Evidence: Document Versus Conversation
A prospective multi-site study analyzing consent processes for pediatric phase I oncology trials provides compelling quantitative evidence of the disparities between consent documents and conversations. The study transcribed and analyzed 69 unique physician-protocol pairs, comparing the word count, Flesch-Kincaid Grade Level (FKGL), and Flesch Reading Ease Score (FRES) of informed consent conversations against their corresponding documents [1].
Table 1: Linguistic Complexity Comparison Between Consent Documents and Conversations
| Parameter | Informed Consent Documents (ICD) | Informed Consent Conversations (ICC) | P-value |
|---|---|---|---|
| Word Count | 6,364 | 4,677 | 0.0016 |
| Flesch-Kincaid Grade Level (FKGL) | 9.7 | 6.0 | <0.0001 |
| Flesch Reading Ease Score (FRES) | 56.7 | 77.8 | <0.0001 |
Table 2: Coverage of Critical Consent Elements During Conversations [1]
| Critical Consent Element | Percentage Discussed During ICC |
|---|---|
| Potential Side Effects | 91% |
| Study Purpose | 86% |
| Safety Mechanisms | 87% |
| Withdrawal Rights | 58% |
| Explanation of Voluntariness | 55% |
| Dosing Cohorts | 44% |
| Dose Escalation | 52% |
| Dose Limiting Toxicities | 26% |
Despite using significantly more understandable language, conversations consistently omitted elements critical to fully informed consent [1]. Investigator experience did not correlate with better coverage of these elements, suggesting that systematic approaches rather than reliance on clinical experience are needed to improve the process.
Experimental Protocols: Methodologies for Consent Process Research
Protocol: Readability Analysis of Consent Components
This methodology details the approach used to generate the quantitative data presented in Section 2, enabling researchers to replicate similar studies in different clinical contexts [1].
Objective: To determine whether length and reading levels of transcribed Informed Consent Conversations (ICCs) are lower than their corresponding Informed Consent Documents (ICDs), and to assess whether investigator experience affects use of simpler language and comprehensiveness.
Materials and Equipment:
- Audio recording equipment
- Transcription software or services
- Microsoft Word 2010 or equivalent software with grammar check functionality
- Readability analysis tools (Flesch-Kincaid Grade Level and Flesch Reading Ease Score)
Procedural Steps:
- Participant Recruitment: Recruit families considering participation in clinical trials at multiple research centers.
- Audio Recording: Audio-record consent conversations between investigators and parents/patients eligible for studies.
- Verification: Transcribe conversations according to a predetermined transcription guide and verify by a second study team member.
- Data Segregation: Isolate investigator-spoken words, excluding questions or words spoken by patients or family members.
- Readability Analysis: Analyze transcribed conversations using grammar check software to collect: (1) word count; (2) Flesch-Kincaid Grade Level (FKGL); and (3) Flesch Reading Ease Score (FRES).
- Document Analysis: Apply identical software analysis to the corresponding IRB-approved informed consent documents.
- Element Coverage Assessment: Code transcripts for coverage of 8 pre-specified critical consent elements through qualitative content analysis.
- Experience Correlation: Correlate years of investigator experience with readability scores and element coverage using appropriate statistical tests (e.g., spearman correlation coefficients, logistic regression).
Validation Method: Use paired-sign test to compare length and readability of transcribed conversations with their corresponding documents.
Protocol: Stakeholder Perception Assessment
This protocol outlines a survey-based methodology for quantifying how clinical research participants and staff experience the informed consent process, with emphasis on contextual factors [5].
Objective: To describe how participants and research staff experience the informed consent process and the contextual factors that contribute to their satisfaction.
Materials and Equipment:
- Anonymous surveys (paper and electronic versions for participants, electronic version for staff)
- Research Electronic Data Capture (REDCap) or equivalent data management tool
- Social media platforms and professional networks for distribution
Procedural Steps:
- Survey Design: Develop separate surveys for research participants (14 multiple choice questions) and research staff (16 multiple choice questions), each with one optional open-ended question.
- Piloting: Pilot surveys among six members of target groups and adjust wording for clarity.
- Participant Recruitment: Distribute surveys to research participants (>18 years old who have taken part in research) via clinical/research teams during routine visits and through digital marketing/social media.
- Staff Recruitment: Distribute staff surveys to those who facilitate informed consent discussions with adult participants via professional networks, newsletters, and social media.
- Data Collection: Collect responses between September 2020 and February 2021.
- Data Management: Input responses into REDCap, with manual verification of data entry accuracy for a random 20% of paper surveys.
- Analysis: Use descriptive statistics for multiple-choice questions and thematic analysis for open-ended responses.
Validation Method: Mitigate acquiescence bias through Likert scales, neutral questions, and "I can't remember" options; minimize social desirability bias by ensuring participants return surveys by post rather than to healthcare teams.
Visualizing the Informed Consent Process
The informed consent process is a multi-stage journey rather than a single event. The following diagram illustrates the key stages and their interrelationships:
This process continues throughout the research relationship, with particular importance placed on providing adequate time for the consent discussion and using techniques like Teach-Back to confirm understanding [5] [2]. Research indicates that consent forms should "begin with a concise and focused presentation of the key information" most likely to assist prospective participants in understanding reasons for or against participation [2] [3].
Research Reagent Solutions: Tools for Effective Consent Processes
Table 3: Essential Resources for Optimizing Informed Consent Processes
| Tool Category | Specific Resource | Function and Application |
|---|---|---|
| Readability Assessment | Flesch-Kincaid Grade Level (FKGL) | Quantifies educational grade level required to understand text [1] |
| Readability Assessment | Flesch Reading Ease Score (FRES) | Measures ease of reading (higher scores reflect easier readability) [1] |
| Regulatory Guidance | 2018 Common Rule §46.116 | Outlines federal requirements for informed consent elements [4] [6] |
| Template Resources | IRB-HSBS Informed Consent Templates | Provides standardized formats including required consent elements [4] |
| Plain Language Tools | PlainLanguage.gov | Offers guidelines for creating clear, understandable consent materials [3] |
| Process Enhancement | Teach-Back Technique | Validates participant understanding through demonstration of knowledge [2] |
| Risk Communication | NCCN Informed Consent Language Database | Provides standardized lay language descriptions of risks [6] |
| Multi-Format Platforms | Electronic Consent (eConsent) | Incorporates dictionaries, animation, videos for different learning styles [7] |
Discussion: Integrating Components for Ethical Practice
The empirical evidence demonstrates that while consent conversations use simpler language than their corresponding documents, they often omit critical elements necessary for fully informed consent [1]. This gap highlights the need for structured approaches that optimize both components of the consent process. Research staff have expressed concerns about participants' understanding of complex information, with 56% of staff worried about comprehension and 63% noting that information leaflets are too long and/or complicated [5].
Successful implementation requires allocating adequate time for consent discussions and considering contextual factors such as setting and timing [5]. The process should be recognized as beginning when potential participants are first educated about the study and continuing through the provision of study results [5] [2]. By integrating plain language principles, appropriate readability tools, and comprehension validation techniques like Teach-Back, researchers can transform informed consent from a bureaucratic hurdle into a meaningful ethical practice that truly respects participant autonomy and promotes understanding.
Application Notes: Integrating Ethical Principles into Practice
Respect for persons is a foundational ethical principle that requires acknowledging the autonomy of individuals and protecting those with diminished autonomy. In practical research terms, this principle is operationalized primarily through the informed consent process, which is more than just a signature on a document—it is a comprehensive process built on trust and respect that continues throughout the study [8] [9].
Foundational Ethical Framework
The National Institutes of Health outlines seven main principles to guide ethical research, several of which directly support respect for persons and autonomous decision-making [8]. These provide a framework for developing application notes that researchers can implement in practice.
Table: Core Ethical Principles Supporting Respect for Persons
| Ethical Principle | Definition | Application to Respect for Persons |
|---|---|---|
| Informed Consent | Potential participants make their own decision about participation based on comprehensive information [8]. | Cornerstone of autonomy; ensures individuals are active agents in the decision to volunteer. |
| Respect for Potential and Enrolled Subjects | Individuals treated with respect from initial approach through study completion, including respecting privacy and right to withdraw [8]. | Acknowledges ongoing autonomy throughout research participation. |
| Favorable Risk-Benefit Ratio | Everything done to minimize risks and maximize benefits for participants [8]. | Demonstrates respect for persons by protecting their well-being. |
| Fair Subject Selection | Recruitment based on scientific goals, not vulnerability or privilege [8]. | Prevents exploitation and promotes equitable respect for all persons. |
Legal and Ethical Foundations of Consent
The legal and ethical validity of consent is rooted in foundational documents including the Nuremberg Code and the Federal Regulations (45 CFR 46.116) [9]. Principle I of the Nuremberg Code states that "the voluntary consent of the human subject is essential," emphasizing that individuals must have sufficient knowledge and comprehension to make an understanding and enlightened decision [9].
For consent to be ethically and legally valid, it must meet three core requirements [9]:
- Information disclosure: Comprehensive explanation of the study purpose, activities, risks, and benefits
- Assessment of competency: Evaluation of the participant's capacity to understand and make a decision
- Voluntariness: Emphasis on the voluntary nature of the decision, free from coercion
Experimental Protocols: Validating Plain Language in Informed Consent
This protocol provides a methodology for empirically testing the effectiveness of plain language requirements in informed consent forms. The study design evaluates comprehension, autonomy perception, and satisfaction across different consent form formats.
Table: Key Variables and Measurement Instruments
| Variable Category | Specific Variables | Measurement Instrument |
|---|---|---|
| Independent Variable | Consent Form Type: Standard vs. Plain Language (8th grade) | Flesch-Kincaid Readability Score [9] |
| Dependent Variables | Comprehension Score | Study-Specific Knowledge Assessment (20 items) |
| Autonomy Perception | Modified Autonomy Preference Index (API) | |
| Decision Satisfaction | Decision Satisfaction Scale (DSS) | |
| Completion Rate | Participation Tracking Log | |
| Demographic Variables | Education level, Health literacy, Age, Prior research experience | Demographic Questionnaire |
Detailed Methodology
Phase 1: Consent Form Development
Ensure both versions contain all required consent elements [9]:
- Purpose of the study
- Study procedures, duration, and tasks
- Reasonably foreseeable risks and discomforts
- Reasonably expected benefits
- Alternative procedures available
- Confidentiality provisions
- Compensation information (if applicable)
- Contact information for questions
- Voluntary participation statement
Validate readability using Flesch-Kincaid Grade Level and Reading Ease formula to confirm the plain language version achieves the target 8th grade reading level [9].
Phase 2: Participant Recruitment and Randomization
- Recruit participants representing the target research population (N=400) with diverse educational backgrounds and health literacy levels.
- Randomly assign participants to one of two experimental conditions:
- Group A: Standard consent form (n=200)
- Group B: Plain language consent form (n=200)
- Obtain preliminary consent for participation in the validation study.
Phase 3: Data Collection
- Administer consent forms to respective groups in controlled settings.
- Allow sufficient time for review (minimum 30 minutes) and provide opportunity for questions.
- Administer assessment battery in the following sequence:
- Comprehension assessment (immediately after consent review)
- Autonomy Perception Index
- Decision Satisfaction Scale
- Demographic questionnaire
Phase 4: Data Analysis
- Compare mean comprehension scores between groups using independent samples t-test.
- Analyze autonomy and satisfaction scores using multivariate analysis of variance (MANOVA).
- Examine moderating effects of demographic variables using regression analysis.
- Calculate effect sizes for all significant differences.
Diagram: Experimental Protocol for Validating Plain Language Consent Forms
The Scientist's Toolkit: Research Reagent Solutions
Table: Essential Materials for Ethical Consent Research
| Tool/Resource | Function/Application | Implementation Example |
|---|---|---|
| Flesch-Kincaid Readability Formula | Quantitatively assesses reading level of consent documents; targets 8th grade level [9]. | Built into Microsoft Word; evaluates sentence length and syllable count. |
| Plain Language Templates | Pre-formatted consent templates with required ethical and regulatory elements [4]. | IRB-HSBS templates include 2018 Common Rule key information elements. |
| Short Form Consent Process | Structured approach for participants with language barriers or limited literacy [10]. | Uses interpreter, witness, and translated documents for non-English speakers. |
| Comprehension Assessment Tools | Validated instruments to measure participant understanding of consent information. | Multiple-choice questions testing key study aspects: procedures, risks, voluntary nature. |
| Informed Consent Guidelines (45 CFR 46.116) | Regulatory framework specifying required consent elements [4] [9]. | Ensures all legally mandated components are included in consent documents. |
Advanced Considerations for Vulnerable Populations
Supporting Autonomous Decision-Making in Vulnerable Groups
Research with vulnerable populations, including people with intellectual and developmental disabilities (IDD), requires additional ethical considerations. The standard approach to respecting autonomous decisions must be enhanced with demonstrated trustworthiness from healthcare providers and researchers [11].
Protocol Modifications for IDD Populations:
- Trustworthiness demonstration: Researchers must proactively establish trustworthiness before assuming capacity for autonomous decisions [11]
- Supported decision-making: Implement frameworks that acknowledge relational autonomy and provide appropriate support
- Dignity of risk: Recognize that the right to make potentially risky decisions is part of personal autonomy and self-determination [11]
Diagram: Supporting Autonomous Decision-Making in IDD Populations
Regulatory Framework and Future Directions
Evolving Standards for Protocol Development
The recently updated SPIRIT 2025 statement provides evidence-based guidance for clinical trial protocols, reflecting methodological advances and emphasizing patient-centered approaches [12]. Key updates include:
- New open science section: Enhancing transparency through trial registration, data sharing, and protocol accessibility [12]
- Patient and public involvement: Explicit requirement describing how patients will be involved in trial design, conduct, and reporting [12]
- Enhanced harm reporting: Additional emphasis on assessment and description of harms [12]
- Expanded checklist: 34 minimum items to address in trial protocols with accompanying explanation and elaboration document [12]
Implementation in Drug Development Context
For professionals in pharmaceutical development, ethical principles must be integrated throughout the clinical trial process. The abbreviated new drug application (ANDA) process demonstrates how ethical considerations are embedded in regulatory frameworks, requiring demonstration of bioequivalence while maintaining ethical standards for human subjects [13].
The evolving landscape of research ethics continues to emphasize the central importance of respect for persons through improved consent processes, enhanced transparency, and meaningful participant engagement. Implementation of these protocols and application notes will strengthen both the ethical conduct and scientific validity of research involving human subjects.
The Revised Common Rule, which took effect in 2019, represents the first significant modernization of the U.S. Federal Policy for the Protection of Human Subjects in nearly three decades [14]. These revisions aim to enhance protections for research participants while reducing unnecessary administrative burdens on investigators [15] [16]. A cornerstone of these changes involves substantial modifications to the informed consent process, with a particular emphasis on improving participant comprehension through new structural and content requirements [17].
The revisions respond to a research environment that has evolved considerably since the original Common Rule's implementation, characterized by an expansion in clinical trial types, increased use of electronic health data, and more sophisticated analytic techniques for studying biospecimens [14]. A key driver for change was documented evidence that consent forms had become excessively lengthy and complex, with one review finding that fewer than one-third of participants adequately understood critical study aspects such as goals, risks, benefits, and randomization procedures [15]. The updated regulations seek to rebalance this dynamic by making consent forms more effective tools for facilitating genuine understanding and autonomous decision-making [18] [17].
Key Regulatory Changes to the Informed Consent Process
Foundational Requirements: The "Reasonable Person" Standard and Key Information
The Revised Common Rule introduced two pivotal general requirements that fundamentally reshape how consent information must be presented to potential research participants:
The Reasonable Person Standard (45 CFR 46.116(a)(4)): Investigators must provide participants with "the information that a reasonable person would want to have in order to make an informed decision about whether to participate, and [an] opportunity to discuss that information" [16]. This standard moves beyond mere technical compliance toward a more participant-centric approach to information disclosure [17].
The Key Information Mandate (45 CFR 46.116(a)(5)): Consent must "begin with a concise and focused presentation of the key information that is most likely to assist a prospective subject or legally authorized representative in understanding the reasons why one might or might not want to participate in the research" [15] [2] [16]. This section must be organized to facilitate comprehension, representing a significant departure from the traditional approach of presenting consent information as a comprehensive list of isolated facts [17].
New and Modified Consent Form Elements
The Revised Common Rule expanded both the basic and additional elements required in informed consent documentation. These changes address evolving ethical concerns in modern research, particularly regarding biological specimens and data usage [15].
Table 1: New Consent Form Elements in the Revised Common Rule
| Element Type | Applicability | Requirement | Rationale |
|---|---|---|---|
| Basic Element | Required when research involves collection of identifiable private information or identifiable biospecimens | A statement indicating whether identifiers may be removed and if the deidentified information/biospecimens may or may not be used or shared for future research [15] [16] | Addresses historical lack of transparency about secondary research uses, as exemplified by the Henrietta Lacks case [15] |
| Additional Element | Required if applicable | Statement on whether biospecimens may be used for commercial profit and if the subject will share in that profit [15] [16] | Discloses potential commercial interests that might otherwise be unknown to participants [17] |
| Additional Element | Required if applicable | Statement on whether clinically relevant research results will be returned to subjects and under what conditions [15] [16] | Clarifies distinction between clinical care and research participation [17] |
| Additional Element | Required if applicable | For research involving biospecimens, statement on whether the research will or might include whole genome sequencing [15] [16] | Informs participants about specific genetic analyses that may raise unique privacy or psychological concerns [15] |
Practical Application: Implementing the Key Information Mandate
Determining What Constitutes "Key Information"
Regulatory guidance suggests that key information should include elements most relevant to a participant's decision-making process. The Secretary's Advisory Committee on Human Research Protections (SACHRP) provides conceptual guidance through key questions investigators should consider when developing this critical section [2]:
Table 2: SACHRP Questions for Determining Key Information
| Question Category | Representative Questions |
|---|---|
| Participation Motivators | What are the main reasons a subject will/will not want to join this study? |
| Study Fundamentals | What research question is the study trying to answer? Why is it relevant to the subject? |
| Participant Experience | What types of activities will subjects do in the research? How will participation impact the subject outside of the research? |
| Novel Aspects | What aspects of participation are likely to be unfamiliar or diverge from expectations? |
While the regulations specify that key information "may include" elements such as voluntary participation, purpose, duration, procedures, risks, benefits, and alternatives, the determination ultimately requires careful consideration of what a reasonable person would need to know to make an informed decision within the specific study context [2].
Protocol for Developing Compliant Consent Forms
Objective: To create an informed consent form that complies with Revised Common Rule requirements while genuinely enhancing participant understanding.
Materials: Study protocol document, regulatory requirements checklist, plain language resources, readability assessment tools.
Procedure:
Preparatory Phase:
Content Outline Development:
Drafting Key Information Section:
- Begin with a concise statement that the activity involves research and participation is voluntary [15].
- Present purpose, expected duration, and procedures in plain language [2].
- Describe reasonably foreseeable risks and discomforts [2].
- Describe any potential benefits to participants or others [2].
- Note appropriate alternative procedures or courses of treatment that might be advantageous [2].
- Limit this section to approximately 1-2 pages using bullet points and white space to enhance readability.
Document Refinement:
- Apply plain language principles throughout the entire document [2] [9].
- Assess reading level using tools like Flesch-Kincaid, aiming for 8th-grade level or lower [9].
- Conduct usability testing with individuals representative of the study population [2].
- Implement teach-back methods where participants explain study elements back to research staff to confirm understanding [2].
Research Reagent Solutions for Consent Development
Table 3: Essential Resources for Developing Compliant Consent Forms
| Tool/Resource | Function | Application Notes |
|---|---|---|
| Regulatory Checklist | Tracks required consent elements under Revised Common Rule | Customize based on study type (e.g., biospecimen research requires additional elements) [15] [16] |
| Readability Software | Assesses reading level using Flesch-Kincaid or similar metrics | Target 8th-grade level or lower; check sentence length and word difficulty [9] |
| Plain Language Thesaurus | Provides alternative common words for complex medical/scientific terms | Replace "antecubital venipuncture" with "blood draw from arm" [2] |
| Usability Testing Protocol | Gathers feedback from individuals representative of study population | Identify confusing sections and improve comprehension before IRB submission [2] |
| Teach-Back Guide | Provides structured approach for research staff to confirm participant understanding | Participants explain key concepts back to staff to verify comprehension [2] |
Discussion: Implications for Research Practice
The Revised Common Rule's emphasis on key information and the reasonable person standard represents a significant shift in the ethical and regulatory approach to informed consent. Rather than treating consent as a static event focused on signature collection, the revised framework conceptualizes consent as an ongoing process centered on participant understanding [17] [9]. This approach acknowledges that merely providing information is insufficient if that information is not comprehended by those making participation decisions [17].
For researchers, these changes require more than simple template modifications. Successful implementation demands a fundamental rethinking of how consent information is structured and presented. The key information section should function as a decision aid rather than a comprehensive study description, emphasizing the most salient points that would influence a person's choice to participate [15] [2]. This approach helps address the problem of information overload, where excessive detail in traditional consent forms often obscured rather than clarified important considerations [18].
The new requirements also present ongoing implementation challenges. The "reasonable person" standard provides limited specific guidance, requiring researchers and IRBs to develop context-dependent interpretations [17]. Similarly, determining what constitutes sufficiently "key" information involves subjective judgment that may vary across study types and populations. These areas will likely evolve through regulatory guidance and practical experience as institutions share their approaches and consent forms become publicly available for certain clinical trials [17].
The Revised Common Rule's key information mandates represent a significant advancement in the ethical conduct of human subjects research. By requiring that consent forms begin with a concise, focused presentation of the most decision-critical information, the regulations aim to make informed consent more meaningful and participant-centered. Successful implementation requires researchers to thoughtfully identify and clearly communicate the factors most relevant to participation decisions, presenting this information in language and formats that facilitate genuine comprehension. When properly executed, these changes have the potential to strengthen the partnership between researchers and participants, enhancing both the ethical integrity of research and public trust in the research enterprise.
The ethical conduct of clinical research is fundamentally grounded in the principle of respect for persons, which is operationalized through the informed consent process [9]. A valid informed consent requires that participants have sufficient knowledge and comprehension of the subject matter to enable an understanding and enlightened decision [9]. However, the effectiveness of this process is critically dependent on one often-overlooked factor: health literacy. Health literacy encompasses the ability to obtain, process, and understand basic health information and services needed to make appropriate health decisions. When research informed consent forms (ICFs) are developed without health literacy principles, they can become a barrier to comprehension rather than a tool for enlightenment. This article explores the critical link between health literacy and comprehension in clinical research, providing data-driven insights and practical protocols for creating accessible informed consent materials.
Quantitative Assessment of Current Consent Form Readability
Recent empirical evidence demonstrates a significant disconnect between the language used in research ICFs and the health literacy levels of potential participants. A 2025 retrospective cross-sectional study assessed the readability of 266 health research ICFs approved by the National Health Research Ethics Committee (NatHREC) in Tanzania, providing crucial quantitative benchmarks for the field [19].
Table 1: Readability Assessment of Health Research Informed Consent Forms (n=266)
| Assessment Metric | Results | Recommended Benchmark | Compliance Status |
|---|---|---|---|
| Page Count | 65.4% had recommended page numbers | ≤3 pages | Moderate |
| Sentence Length | 81.6% had longer sentences | ≤15 words per sentence | Poor |
| Readability (Flesch Reading Ease) | 80.5% were difficult to read | Score of 60-70 (Standard) | Poor |
| Reading Grade Level | Required US Grade 10 (Form Four in Tanzania) | ≤US Grade 8 | Poor |
The study employed two validated readability formulas: Flesch Reading Ease (FRE) and Flesch-Kincaid Readability Grade Level (FKRGL) [19]. Statistical analysis revealed that sentence length showed a significant correlation with difficult reading levels (p-values < 0.001 at 95% confidence level), indicating that verbose sentence construction is a primary contributor to poor comprehension [19]. These findings align with challenges observed in diverse global contexts, including Iran, the USA, Ireland, and several African nations [19].
Experimental Protocols for Readability Assessment and Improvement
Protocol 1: Readability Testing for Informed Consent Forms
Purpose: To quantitatively and qualitatively assess the readability of research ICFs using standardized metrics and human feedback.
Materials:
- ICF in editable digital format (Microsoft Word)
- Flesch-Kincaid readability software (built into Microsoft Word)
- Participant recruitment from target population
- Assessment checklist
- Structured interview guide
Methodology:
- Document Preparation: Convert ICF to Microsoft Word format. Remove all identifying information (investigator names, institutional logos) to prevent bias [19].
- Automated Readability Analysis: a. Open the document in Microsoft Word b. Navigate to File > Options > Proofing c. Select "Show readability statistics" d. Run spelling and grammar check to trigger readability report e. Record FRE and FKRGL scores, word count, and sentence length [19]
- Human Comprehension Assessment: a. Recruit 5-10 participants representative of the target research population b. Present the ICF using the planned consent process c. Administer a comprehension assessment using the teach-back method d. Conduct structured interviews to identify problematic phrases and concepts
- Data Synthesis: Combine quantitative readability scores with qualitative feedback to identify specific areas for improvement.
Validation: Compare final readability scores against benchmark of ≤8th grade reading level and FRE score of 60-70 [4] [9].
Protocol 2: Plain Language Revision of Informed Consent Forms
Purpose: To systematically transform technically complex ICFs into plain language documents that maintain regulatory compliance while enhancing comprehension.
Materials:
- Source ICF with readability assessment data
- Plain Language Checklist [20]
- Regulatory requirements checklist [2]
- Multidisciplinary team (investigator, plain language expert, community representative)
Methodology:
- Requirement Mapping: a. Create a table of all legally required consent elements per 45 CFR 46.116 [2] [4] b. Identify state-specific and industry-specific additional requirements c. Categorize elements as "key information" versus "additional information"
- Key Information Section Development:
a. Begin with a concise presentation of key information as required by the Revised Common Rule [2]
b. Structure key information using SACHRP guiding questions [2]:
- What are the main reasons a subject will want to join this study?
- What are the main reasons a subject will not want to join this study?
- How will the subject's experience differ from standard treatment?
- Plain Language Implementation: a. Apply the "Everyday Words for Public Health Communication" principles [20] b. Reduce sentence length to average 15-20 words [21] c. Use active voice and second person ("you") throughout [4] d. Organize content into logical chunks with descriptive headings [20] e. Replace technical jargon with common, everyday alternatives f. Incorporate visual elements, bullet points, and white space to improve readability
- Validation and Refinement: a. Conduct usability testing with the target population b. Use teach-back methods to verify comprehension [2] c. Obtain IRB/EC review and approval
Quality Control: Apply the Plain Language Checklist to ensure all principles have been implemented [20].
Table 2: Research Reagent Solutions for Health Literacy Implementation
| Tool Name | Source | Function | Application Context |
|---|---|---|---|
| Flesch-Kincaid Readability Formulas | Microsoft Word | Quantifies reading grade level and ease of reading | Automated initial assessment of ICF readability [19] [9] |
| Plain Language Checklist | CDC | Provides criteria for clear communication | Guiding document revision and quality assurance [20] |
| Everyday Words for Public Health Communication | CDC | Thesaurus for simplifying technical terms | Replacing jargon with common alternatives [20] |
| Informed Consent Templates | Institutional Review Boards | Regulatory-compliant ICF structure | Ensuring all required elements are included [4] |
| Teach-Back Method | Health Literacy Tools | Verbal verification of understanding | Assessing participant comprehension during consent process [2] |
The Three Pillars of Effective Consent: A Framework for Implementation
The development of literacy-appropriate consent forms requires addressing three foundational pillars: Purpose, Audience, and Process [2]. This framework ensures both regulatory compliance and genuine participant understanding.
Purpose-Driven Consent Design
Informed consent serves multiple purposes beyond regulatory compliance. Effective ICFs must simultaneously function as decision-making aids for potential participants, educational tools for research staff, ethical frameworks for IRB reviewers, and reference documents for enrolled participants [2]. This multidimensional purpose necessitates careful consideration of what information to include and how to present it clearly. The fundamental goal remains facilitating understanding to support autonomous decision-making about research participation [2].
Audience-Centered Communication Strategies
Understanding the target audience is critical for developing effective consent materials. This requires consideration of demographic factors, cultural backgrounds, medical contexts, and literacy levels [2]. Best practices include involving people from the study population during protocol and consent form development to ensure materials meet their informational needs and are designed for a study they would want to join [2]. For multicultural contexts, translation into relevant languages and adaptation of cultural references may be necessary [22].
Process-Oriented Consent Implementation
Consent is an ongoing process rather than a single form-signing event [9]. This process begins with initial study education and continues throughout the research participation period [2]. Key considerations include determining when and how participants will receive consent forms, how long they will have to review them, who will be available to answer questions, and how the consent process will support ongoing questioning and discussion [2]. The most effective processes incorporate interactive elements, particularly the evidence-based teach-back method where participants demonstrate understanding by explaining concepts in their own words [2].
Regulatory Framework and Key Information Requirements
The Revised Common Rule (2018) mandates that consent forms "begin with a concise and focused presentation of the key information that is most likely to assist a prospective subject or legally authorized representative in understanding the reasons why one might or might not want to participate in the research" [2] [4]. This regulatory requirement emphasizes the importance of foregrounding the most critical elements for decision-making.
Table 3: Key Information Elements for Informed Consent Forms
| Element Number | Required Element | Application Notes | Regulatory Reference |
|---|---|---|---|
| 1 | Statement that project is research and participation is voluntary | Must be explicit and prominent | 45 CFR 46.116 [4] |
| 2 | Summary of research (purpose, duration, procedures) | Focus on what participants will experience | 45 CFR 46.116 [4] |
| 3 | Reasonably foreseeable risks or discomforts | Describe in specific, understandable terms | 45 CFR 46.116 [4] |
| 4 | Reasonably expected benefits | Distinguish between direct and societal benefits | 45 CFR 46.116 [4] |
| 5 | Alternative procedures or course of treatment | Primarily for clinical research | 45 CFR 46.116 [4] |
The Secretary's Advisory Committee on Human Research Protection (SACHRP) provides guiding questions to help determine what constitutes key information for specific studies, including: "What are the main reasons a subject will want to join this study?" and "What are the main reasons a subject will not want to join this study?" [2]. This approach ensures that the key information section addresses the most salient considerations for potential participants.
The critical link between health literacy and comprehension in clinical research demands systematic attention throughout the informed consent development process. Quantitative evidence demonstrates that most current consent forms exceed recommended readability levels, creating barriers to understanding [19]. By implementing structured protocols for readability assessment, applying plain language principles grounded in regulatory requirements, and adopting a comprehensive framework addressing purpose, audience, and process, researchers can develop consent materials that truly support autonomous decision-making. These approaches not only fulfill ethical obligations but also enhance research quality by ensuring participants genuinely understand what their involvement entails.
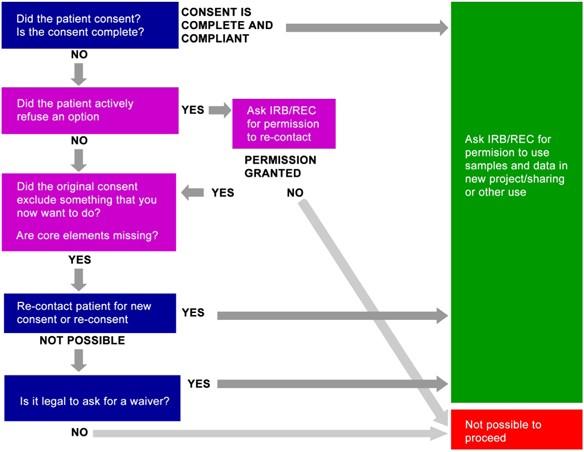
A valid informed consent process is a cornerstone of clinical research, essential for preserving respect for participant autonomy and dignity [23]. However, in specific and narrowly defined research contexts, such as emergency care and certain pragmatic trials, obtaining prospective informed consent is not always possible. For patients presenting with emergent, life-threatening conditions, the acuity of their illness and time-sensitive need for intervention often render them incapable of providing meaningful prospective consent [24] [25]. Similarly, some pragmatic trials embedded within routine healthcare systems encounter practical obstacles that make individual consent for data use or intervention impractical.
To address these challenges, regulations permitting an exception from informed consent (EFIC) or a waiver of informed consent (WIC) were established. These frameworks create a narrow pathway for ethically conducting vital research that would otherwise be impossible, while implementing robust alternative protections for participants [24] [25]. This document provides detailed application notes and experimental protocols for the ethical application of EFIC and WIC, framed within a broader thesis on informed consent and the critical role of plain language.
Regulatory Frameworks and Quantitative Review
The foundational regulations for EFIC and WIC were developed in 1996. EFIC is detailed in the U.S. Food and Drug Administration (FDA) regulation 21 CFR 50.24, while WIC is described in the Department of Health and Human Services (DHHS) regulation 45 CFR 46.101 [24] [25]. These are not interchangeable; EFIC applies specifically to FDA-regulated emergency research, whereas WIC applies to a broader category of minimal-risk research under the Common Rule.
A 20-year review of the use of these regulations provides critical quantitative insight into their application, summarized in the table below.
Table 1: Twenty-Year Review of EFIC/WIC Study Characteristics (28 Completed Studies) [24] [25]
| Review Category | Findings | Frequency (n, %) |
|---|---|---|
| Type of Consent Used | Exception from Informed Consent (EFIC) | 24 (86%) |
| Waiver of Informed Consent (WIC) | 4 (14%) | |
| Common Pathologies Studied | Cardiac Arrest | 10 |
| Hemorrhagic Shock | 6 | |
| Traumatic Brain Injury | 5 | |
| Reported Pre-Study Requirements | FDA IND/IDE Application | 14 (50%) |
| Community Consultation | 13 (46%) | |
| Public Disclosure | 10 (36%) | |
| IRB-Requested Opt-Out Procedures | 7 (25%) |
This review highlights that while these regulations have enabled progress in treating devastating conditions, reporting of key ethical safeguards is inconsistent. Just 46% of publications explicitly justified the need for using EFIC/WIC, indicating a significant area for improvement in transparent reporting [24] [25].
Ethical Foundations and Tensions
The use of EFIC/WIC creates an inherent ethical tension between competing moral imperatives. This tension lies between the duty to respect participant autonomy and the compelling societal need to generate evidence to improve outcomes for populations with acute, life-threatening conditions [23]. In emergency obstetric and newborn care in low-resource settings, for example, this tension is acute; prohibiting research denies a vulnerable population access to potential research benefits, while enrolling without consent risks violating autonomy [23].
The ethical justification for these exceptions rests on a balance of principles:
- Beneficence: The potential to provide life-saving therapies and advance knowledge for future patients.
- Non-maleficence: The requirement to minimize risks and protect participants from harm.
- Justice: The fair inclusion of populations who suffer from acute conditions, ensuring they are not denied the benefits of research participation [23].
The diagram below illustrates the logical pathway and ethical balancing test that must be satisfied to justify a waiver or exception of consent.
Application Notes and Experimental Protocols
This section provides a detailed methodology for implementing the key ethical and regulatory requirements for EFIC research.
Protocol for Community Consultation and Public Disclosure
Community consultation and public disclosure are not merely regulatory checkboxes but are fundamental processes for respecting the autonomy of the community from which participants will be enrolled.
Table 2: Protocol for Community Consultation and Public Disclosure
| Component | Objective | Detailed Methodology | Outcome Measures |
|---|---|---|---|
| Stakeholder Mapping | Identify groups representing the community at risk for the medical condition. | - Review epidemiological data for the condition.- Identify patient advocacy groups, community leaders, and clinicians in the field.- Include past patients and their families. | A list of relevant organizations and leaders to engage. |
| Consultation Methods | Gather diverse perspectives on the research study's acceptability. | - Conduct public meetings/webinars.- Organize focus groups.- Deploy structured surveys (online, telephone, in-clinic).- Present the study's purpose, risks, benefits, and why EFIC is necessary. | Qualitative feedback and quantitative survey data on the perceived acceptability of the study. |
| Public Disclosure | Inform the broader community about the research. | - Issue press releases to local media.- Publish information on hospital/clinic websites and social media.- Display informational posters in relevant clinical areas (e.g., ED, ICUs). | Documentation of the methods used, materials distributed, and the reach of the disclosure. |
| Reporting to IRB/FDA | Demonstrate compliance and incorporate feedback. | - Summarize the consultation process, feedback received, and how the protocol was modified in response.- Provide copies of all disclosure materials. | A final report submitted to the overseeing IRB and, if applicable, the FDA. |
Protocol for Implementing Opt-Out Procedures
The opt-out mechanism is a critical safeguard that provides a direct avenue for individuals to refuse potential enrollment in advance, thereby preserving a element of individual choice.
Experimental Workflow:
- Opt-Out Mechanism Design: The IRB-approved mechanism must be accessible and understandable. This typically involves a dedicated 24/7 website and a toll-free telephone number. All public disclosure materials must clearly display these opt-out channels.
- Data Management System: Establish a secure, encrypted database to record individuals who opt out. The system must capture the individual's name and contact information, the date of opt-out, and a unique identifier.
- Integration with Enrollment Process: Prior to enrolling any patient under EFIC, the research team must check this secure database. The check must be documented in the study records.
- Respecting Opt-Out: If an individual is found in the database, they must not be enrolled in the study under any circumstances, even if they otherwise meet all inclusion criteria. Their clinical care will proceed with the local standard of care.
The workflow for checking and respecting an opt-out request during a potential enrollment scenario is detailed below.
The Scientist's Toolkit: Essential Reagents and Materials
For researchers designing and conducting trials under EFIC/WIC, the following "reagents" are essential.
Table 3: Essential Research Reagent Solutions for EFIC/WIC Trials
| Item / Solution | Function / Purpose | Specific Application Notes |
|---|---|---|
| FDA Guidance Document on EFIC | Provides the definitive regulatory framework for designing EFIC studies. | Used to ensure all pre-study requirements (community consultation, public disclosure, IRB oversight) are met. Essential for protocol development. |
| Institutional Review Board (IRB) | Provides independent ethical oversight and approval. | The IRB must be specially constituted and familiar with EFIC regulations. They review and approve the entire plan for community consultation, opt-out procedures, and the study protocol itself. |
| Community Consultation Toolkit | A structured set of materials and methods for engaging the community. | Includes survey templates, focus group guides, and presentation slides. Its function is to operationalize the ethical principle of community respect and gather meaningful feedback. |
| Secure Opt-Out Database | A dedicated, secure IT system for managing opt-out requests. | This system is critical for implementing the key safeguard of individual refusal. It must be HIPAA-compliant, accessible 24/7, and integrated into the enrollment workflow. |
| Plain Language Summary Template | A tool for creating accessible public disclosure and participant-facing materials. | Based on WHO templates that recommend a 6th-8th grade reading level [26]. Its purpose is to ensure that information about the trial is truly understandable to the public, aligning with core principles of informed consent even when consent is waived. |
Integration with Plain Language Principles
Even in research where consent is waived, the ethical obligation to communicate clearly and transparently remains paramount. The 2018 revision to the Common Rule mandates that informed consent forms begin with a "concise and focused presentation of key information" to facilitate comprehension [27]. This principle must extend to all public-facing documents in an EFIC/WIC study, including community consultation materials and public disclosure notices.
Best Practices for Plain Language in EFIC Contexts:
- Readability: Use tools like the Flesch-Kincaid test to ensure materials are written at a 6th to 8th-grade reading level [26] [28]. The World Health Organization recommends this level for participant materials [26].
- Clarity and Structure: Avoid jargon and technical terms. Use active voice, short sentences, and visual aids like timelines and flowcharts to explain study procedures [28].
- Actionability: Ensure that information about how to opt-out is presented clearly and prominently, allowing individuals to easily understand and act upon their right to refuse participation.
Emerging technologies, including Large Language Models (LLMs), show promise in automating the generation of more readable and actionable consent-related documents. One recent study demonstrated that LLM-generated consent forms significantly improved readability, understandability, and actionability scores compared to human-generated forms, without sacrificing accuracy [27]. This suggests a future where plain language is more scalably achieved, even in complex trial settings.
The regulations for exception from and waiver of informed consent provide a vital, ethically defensible pathway for conducting research in emergency and pragmatic settings where traditional consent is not feasible. The successful and ethical application of these regulations hinges on a steadfast commitment to their foundational safeguards: meaningful community consultation, transparent public disclosure, and functional opt-out mechanisms. Furthermore, integrating the principles of plain language into all public communications is not optional but a core ethical requirement, ensuring that the spirit of informed consent—respect for persons and their autonomy—is upheld even when the formal consent process is waived. As research methodologies and technologies evolve, so too must our commitment to these protective practices, ensuring that the pursuit of life-saving knowledge never outstrips our duty to protect the individuals and communities we serve.
The How: A Step-by-Step Framework for Writing and Designing Accessible Forms
A well-defined preparatory phase is the cornerstone of a valid and ethical informed consent process in clinical research. This initial step ensures that the consent is not merely a regulatory formality but a meaningful, ongoing dialogue that respects participant autonomy. For research to be ethical, participants must willingly volunteer, having been adequately informed about the study [4]. This protocol outlines the critical preparatory actions—defining the study's purpose, understanding the target audience, and designing the consent process—within the context of evolving global standards, including the ICH E6(R3) guidelines effective in 2025, which emphasize a principles-based and risk-proportionate approach [29]. Adherence to these protocols is essential for protecting participant rights, ensuring regulatory compliance, and generating reliable scientific data.
Defining the Research Purpose and Key Information
The first preparatory action is to articulate a clear and concise summary of the research that can be easily communicated to a potential participant. This involves distilling complex scientific objectives into key information that facilitates understanding and supports a decision about participation.
Regulatory Framework for Key Information
The revised Common Rule (2018) and harmonizing FDA guidance mandate that informed consent documents begin with a "concise and focused" presentation of key information [4] [30]. This section must help potential participants understand why they might or might not want to be part of a research study. The ICH E6(R3) guideline further reinforces the need for clear communication as part of its core ethical principles [29].
The Five Key Information Elements
Investigators must prepare a summary that addresses the following five core elements, which form the basis of the key information section [4]:
Table 1: Core Elements of Key Information in Informed Consent
| Element Number | Element Description | Application Notes |
|---|---|---|
| 1 | A statement that the project is research and participation is voluntary. | A foundational declaration of the activity's nature and the individual's right to choose. |
| 2 | A summary of the research, including: purpose, duration, and list of procedures. | Provides a high-level overview of what the study entails and the participant's commitment. |
| 3 | Reasonable, foreseeable risks or discomforts. | Critical for a realistic understanding of potential physical, psychological, or social harms. |
| 4 | Reasonable, expected benefits. | Must clearly distinguish direct benefits to participants from benefits to society; compensation is not a benefit. |
| 5 | Alternative procedures or course of treatment, if any. | Primarily applies to clinical trials of therapeutic interventions. |
The logical flow from establishing the research context to outlining participant-specific implications can be visualized in the following workflow:
Identifying and Analyzing the Target Audience
A one-size-fits-all approach to consent is ineffective. The consent process and documents must be tailored to the specific subject population to ensure comprehension and genuine informed consent.
Audience Analysis and Plain Language
Informed consent documents must be written in plain language at a level appropriate to the subject population, generally at an 8th-grade reading level [4] [9] [31]. This requires avoiding technical jargon and using straightforward, understandable language. The use of complex scientific and medical terms should be avoided in favor of explanations the general public can understand [31]. Tools like the Hemingway Readability Checker or the Flesch-Kincaid Grade Level formula can help assess the reading level of consent materials [9] [31].
Considerations for Vulnerable Populations
Certain populations are deemed vulnerable and require additional safeguards during the consent process. Researchers must identify if their study involves such groups and plan for enhanced protections [4] [9].
Table 2: Audience Analysis and Protections Planning
| Audience Category | Key Considerations | Required Protections & Adaptations |
|---|---|---|
| General Adult Population | Aim for ≤8th grade reading level; use second-person ("you") language [4] [32]. | Standard plain language principles; use of visuals/infographics to aid understanding [31]. |
| Children | Legal capacity to consent is absent; assent is required. | Must first obtain permission of parents in addition to the consent of the children [4]. |
| Cognitively Impaired Individuals | Assessment of capacity to understand and consent is needed [9]. | Consent from a legally authorized representative; assessment of capacity to consent may be required [31]. |
| Non-English Speaking Populations | Consent documents and process must be linguistically accessible. | Use of translated consent forms or a short-form consent process with an interpreter [31]. |
| Other Vulnerable Groups (e.g., prisoners, employees) | Potential for coercion or undue influence [9]. | IRB review of additional parameters for recruitment and consent to minimize perceived coercion [31]. |
The process for assessing the target audience and implementing appropriate consent strategies is outlined below:
Designing the Informed Consent Process
Informed consent is a process, not a single event centered on a signature [9]. It is built on trust and respect, spread throughout a study, beginning before data collection and continuing through the participant's involvement.
The Consent Process Workflow
The process typically involves a structured conversation, followed by documentation and ongoing engagement.
- Pre-Consent Preparation: The research team must confidently know and understand the study activities and prepare for common questions. They should allocate sufficient time for the consent conversation and be prepared to explain complex concepts in simple, culturally sensitive terms [9].
- The Initial Conversation: This is an open communication exchange free of coercion and bias. It involves discussing the key information and all required elements of consent, allowing the prospective participant ample time to ask questions [9] [31].
- Assessment of Comprehension: Researchers should check in with participants throughout the process to ensure understanding, particularly regarding risks and benefits. Techniques like the "teach-back" method, where participants explain the information back in their own words, can be highly effective [31].
- Documentation: After the conversation and once all questions are answered, the participant signs the consent form. This documentation can be a traditional written signature or an eConsent via an approved digital platform, a practice explicitly supported by the media-neutral language of ICH E6(R3) [31] [29].
- Ongoing Consent: The process continues by providing new information that arises during the trial, re-consenting participants if necessary, and continually affirming the participant's willingness to continue [9].
Experimental Protocol: Implementing the Teach-Back Method
The teach-back method is a validated technique to ensure participant comprehension during the consent conversation.
- Objective: To verify the participant's understanding of key study elements, such as purpose, procedures, risks, benefits, and the voluntary nature of participation.
- Materials: Informed Consent Form, protocol summary.
- Methodology:
- After explaining a key concept (e.g., the main procedures of the study), the investigator asks the participant to explain it back in their own words. Example: "To make sure I explained everything clearly, could you please tell me in your own words what you'll be asked to do during the study?"
- The investigator listens carefully for accuracy and completeness.
- If the understanding is incorrect or incomplete, the investigator re-explains the information using different, clearer language.
- Steps 1-3 are repeated until the participant demonstrates a clear understanding of the concept.
- This process is applied to all critical aspects of the study, particularly those relating to risks and the rights of a volunteer.
- Documentation: The use of the teach-back method and the general assessment of participant understanding should be noted in the study records.
Preparing a robust consent process requires leveraging specific tools and resources to ensure clarity, compliance, and participant understanding.
Table 3: Essential Research Reagent Solutions for Consent Process Development
| Tool/Resource | Function | Application Example |
|---|---|---|
| Institutional Consent Template (e.g., IRB-HSBS template) | Provides a pre-formatted structure containing all required regulatory and institutional consent elements [4]. | Ensures new consent documents comply with 2018 Common Rule and institutional policy, preventing omission of critical clauses. |
| Readability Analyzer (e.g., Hemingway App, Flesch-Kincaid) | Quantitatively assesses the reading grade level of written text [9] [31]. | Used to revise and simplify consent form language to achieve the target ≤8th-grade reading level. |
| Plain Language Thesaurus (e.g., MRCT Center Toolkit) | Provides simpler, lay-language alternatives for complex scientific and medical terms [31] [6]. | Replaces jargon like "hypotension" with "low blood pressure" to improve participant comprehension. |
| Digital Health Technology (DHT) & eConsent Framework | Guides the integration of digital tools for remote consenting and data collection, as per ICH E6(R3) [31] [29] [6]. | Implmenting a REDCap eConsent framework with embedded multimedia (videos, infographics) to explain complex procedures. |
| Informed Consent Language (ICL) Database (e.g., from NCCN) | Offers standardized lay-language descriptions of risks and events common in clinical research [6]. | Drafting clear, consistent descriptions of chemotherapy side effects in an oncology trial consent form. |
Regulatory and Ethical Framework
The preparatory phase is conducted within a strict regulatory landscape. Key U.S. regulations include the HHS (45 CFR 46) and FDA (21 CFR 50) rules [4] [33] [32]. A significant development for 2025 is the harmonization between FDA and OHRP (Office for Human Research Protections) guidance on the requirement for a "key information" section, which aligns standards for federally funded and drug/device trials [30]. Ethically, the process is grounded in the Belmont Report principles of respect for persons, beneficence, and justice, requiring researchers to ensure participation is voluntary and informed, without exculpatory language [9] [32]. The upcoming ICH E6(R3) guideline reinforces these principles, promoting a flexible, quality-focused approach adaptable to diverse trial designs and technologies [29].
Regulatory Framework and Essential Elements
Informed consent is a foundational ethical and regulatory requirement in human subjects research. The process involves providing potential participants with key information about a study to enable a voluntary decision on whether to participate [34]. The following table summarizes the core elements required by major U.S. regulatory bodies.
Table 1: Essential Elements of Informed Consent per U.S. Regulations
| Element Description | Common Rule (45 CFR 46) [35] | FDA Regulations (21 CFR 50) [35] | Plain Language Application |
|---|---|---|---|
| Statement that study involves research | Required | Required | Clearly state "This is a research study." |
| Explanation of study purpose | Required | Required | Explain why the research is being done in 1-2 sentences. |
| Description of procedures | Required | Required | List all procedures in order, using common terms. |
| Description of foreseeable risks | Required | Required | Group risks by likelihood and severity; use descriptive scales. |
| Description of any benefits | Required | Required | Differentiate between direct benefits to participants and societal benefits. |
| Disclosure of alternative procedures | Required | Required | List standard treatments available outside the study. |
| Description of confidentiality | Required | Required | Explain who will see the information and how it will be protected. |
| Explanation of compensation | Required for > Minimal Risk | Required for > Minimal Risk | State clearly if no compensation is provided for injury. |
| Contact information for questions | Required | Required | Provide names and phone numbers of key contacts. |
| Statement that participation is voluntary | Required | Required | Use explicit language: "Your participation in this study is completely voluntary." |
Experimental Protocol for Informed Consent Document Development
Protocol: Systematic Development of a Plain Language Consent Form
Objective: To create a legally compliant informed consent document that is understandable to participants at an 8th-grade reading level.
Materials:
- Regulatory templates (e.g., from institutional IRB library) [34]
- Word processing software with Flesch-Kincaid readability tool
- 12-point Arial or Times New Roman font
Methodology:
- Template Selection: Obtain and utilize the most current IRB-approved consent form template from your institution's repository to ensure all structural elements are present [34].
- Content Drafting: Insert study-specific information into the template using complete sentences. Adhere to the following guidelines:
- Use short, simple, and direct sentences.
- Spell out all abbreviations and acronyms upon first use.
- Avoid technical jargon and scientific terms. If necessary, define them in simple language [34].
- Readability Assessment: Utilize the Flesch-Kincaid readability tool within your word processor to measure the document's grade level. Refine the language until an 8th-grade reading level is achieved [34].
- Formatting and Layout:
- Maintain a 12-point or larger font size.
- Ensure adequate white space and clear section headings.
- Use line numbers during the review process, which should be removed after IRB approval [34].
- Regulatory Review and Approval: Submit the drafted consent document to the Institutional Review Board (IRB) for review and approval before use in any participant enrollment [35].
Experimental Protocol for Consent Process Assessment and Validation
Protocol: Quantitative and Qualitative Assessment of Consent Comprehension
Objective: To evaluate participant understanding of the informed consent document and process through mixed-methods analysis.
Materials:
- Approved informed consent document
- Digital data collection platform (e.g., survey host)
- Consent comprehension questionnaire
- Secure data storage system
Methodology:
- Participant Enrollment: Recruit participants according to the study's IRB-approved protocol.
- Consent Process: Conduct the informed consent process as approved by the IRB.
- Data Collection:
- Quantitative Data: Administer a structured comprehension questionnaire post-consent. The questionnaire should use multiple-choice and true/false questions to assess understanding of key study elements (e.g., purpose, procedures, risks, rights) [36].
- Qualitative Data: For a subset of participants, conduct brief, semi-structured interviews or solicit open-ended feedback to gather in-depth context on the clarity of the consent document and identify any persistent areas of confusion [36].
- Data Analysis:
- Quantitative Analysis: Calculate cumulative comprehension scores and item-specific correct response rates. Summarize data using descriptive statistics (e.g., means, standard deviations, percentages) [36].
- Qualitative Analysis: Transcribe and code interview responses. Identify common themes and specific phrases or concepts that participants found unclear [36].
- Iterative Refinement: Use the combined quantitative and qualitative findings to revise and improve the consent document and process, submitting changes to the IRB for approval as necessary.
Table 2: Data Collection and Analysis Matrix for Consent Comprehension
| Data Type | Collection Method | Metric | Analysis Technique | Output |
|---|---|---|---|---|
| Quantitative | Comprehension Questionnaire | Correct Response Rate (%) | Descriptive Statistics (Mean, SD) | Identifies gaps in understanding of key facts [36]. |
| Quantitative | Comprehension Questionnaire | Total Comprehension Score | Descriptive Statistics (Mean, SD) | Provides an overall measure of document effectiveness [36]. |
| Qualitative | Semi-structured Interviews | Participant Quotes & Feedback | Thematic Analysis & Coding | Reveals reasons behind misunderstandings and context for numerical data [36]. |
| Integrated | Combined Quantitative & Qualitative | Points of Convergence/Divergence | Comparative Analysis | Creates a unified report for actionable revisions, telling the complete story of comprehension [36]. |
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Essential Materials for Informed Consent Process and Documentation
| Item | Function/Application |
|---|---|
| IRB-Approved Consent Template | Provides a pre-formatted structure that includes all required regulatory elements, streamlining document creation and ensuring compliance [34]. |
| Readability Assessment Software | Objectively measures the grade level of written text to enforce plain language standards (e.g., targeting an 8th-grade reading level) [34]. |
| Electronic Signature System | Enables remote consenting and provides a legally valid method for obtaining and documenting participant consent electronically [34]. |
| Debriefing Script Template | Provides a standardized framework for explaining any deception or incomplete disclosure used in the study and for informing participants of their data rights post-study [34]. |
| Data Anonymization Toolset | Software or procedures used to remove or code personal identifiers from research data, fulfilling confidentiality promises made in the consent document [35]. |
Visualization of Informed Consent Development Workflow
The use of plain language in informed consent forms (ICFs) is an ethical and practical imperative in clinical research. Effective communication ensures that participants fully comprehend the nature of the research, potential risks and benefits, and their rights, thereby enabling truly informed decision-making. Jargon—specialized technical language familiar only to experts—creates a significant barrier to understanding for research participants. Federal guidelines stipulate that language in ICFs should be comprehensible to subjects at the 8th-grade reading level [37]. This application note provides a structured framework for researchers to systematically identify and replace jargon with simple, clear alternatives, enhancing participant understanding and upholding the ethical standards of the informed consent process. Studies demonstrate that improving the readability of ICFs significantly increases participant comprehension and satisfaction [38].
Quantitative Data Presentation: Jargon and Plain Language Equivalents
The following tables provide a structured reference for replacing common jargon terms with their plain language equivalents, categorized for easy application.
Table 1: General Medical and Procedural Jargon
| Jargon Term | Plain Language Alternative | Rationale for Replacement |
|---|---|---|
| Abrasion | Scraped skin/area where skin is scraped away [37] | Uses concrete, familiar words. |
| Acute | New, recent, or sudden [37] | Avoids clinical ambiguity. |
| Adverse Effect | Unwanted effect [37] | Simpler, more direct terminology. |
| Analgesic | Drug used to control pain [37] | Defines the term by its function. |
| Benign | Not cancer, usually without serious consequences [37] | Provides context and reassurance. |
| Bilateral | On both sides of the body [37] | Clear anatomical description. |
| Biopsy | Removing a small piece of tissue for lab testing [37] | Describes the action and purpose. |
| Chronic | Lasting a long time [37] | Contrasts clearly with "acute." |
| Contusion | Bruise [37] | Uses a common, everyday word. |
| Edema | Swelling from fluid build-up [37] | Describes the visible condition. |
Table 2: Research-Specific and Statistical Jargon
| Jargon Term | Plain Language Alternative | Rationale for Replacement |
|---|---|---|
| Clinical Trial | A research study with patients [37] | Clarifies the nature of the activity. |
| Control | A standard treatment or procedure used for comparison [37] | Explains the concept of comparison. |
| Contraindications | Medical reasons that prevent you from using a certain drug [37] | Focuses on patient applicability. |
| Double-blind trial | A study where neither you nor the research staff know which treatment you are receiving [37] | Explains the mechanism simply. |
| Efficacy | How well something works [37] | A succinct, clear alternative. |
| Randomization | You will be put into a study group by chance, like flipping a coin [37] | Uses a familiar analogy. |
| Informed Consent | A process to learn about the study and agree to take part [38] | Defines the process in active terms. |
Experimental Protocol: Readability Assessment and Jargon Replacement Workflow
This protocol outlines a systematic, step-by-step methodology for evaluating and revising an informed consent form to improve its readability by removing jargon and simplifying language.
Materials and Reagents
Table 3: Research Reagent Solutions for Readability Analysis
| Item | Function/Application |
|---|---|
| Original Informed Consent Form | The document to be analyzed and revised. |
| Digital Text File (.docx, .txt) | For software-based readability analysis. |
| Readability Assessment Software | Automated tools to calculate readability scores and identify complex language. |
| Validated Plain Language Glossary | A reference for replacing jargon with simple terms [37]. |
| Style Guide | A guide for using active voice and short sentences. |
Step-by-Step Procedure
Initial Readability Assessment:
- Create a digital copy of the ICF text, removing any personal identifiers.
- Input the text into readability analysis software.
- Record the initial readability scores, including Flesch-Kincaid Grade Level, sentence length, and syllable count. The initial analysis of a sample form may indicate it is suitable only for persons with a university degree [38].
Jargon Identification and Replacement:
- Perform a manual review of the ICF, highlighting all technical and medical jargon.
- Cross-reference each identified jargon term with a validated plain language glossary [37].
- Systematically replace each jargon term with its plain language equivalent, as detailed in Table 1 and Table 2.
Structural and Syntactic Simplification:
- Break down long, complex sentences into shorter ones (aim for ≤ 15 words).
- Change passive voice constructions to active voice (e.g., "You will be given..." instead of "The drug will be administered to you...").
- Use second-person pronouns ("you") to address the participant directly [38].
- Ensure the document uses a logical flow with clear headings and bullet points.
Post-Revision Readability Assessment:
- Re-analyze the revised ICF text using the same readability software.
- Document the new readability scores. A successful revision should lower the required reading grade level significantly, for example, from a university level to a 4th-6th-grade level [38].
Validation and Feedback:
- Present the revised ICF to a small panel of individuals representing the target participant population.
- Use questionnaires or interviews to assess comprehension and clarity.
- Incorporate feedback to make final revisions to the document.
Visualization of Jargon Replacement Workflow
The following diagram illustrates the logical sequence and iterative nature of the protocol described above.
Table 4: Key Resources for Creating Plain Language Informed Consent Forms
| Resource | Function | Application in ICF Development |
|---|---|---|
| MedLine Plus | Provides simplified explanations of medical procedures and equipment [37] | Reference for creating accurate, easy-to-understand descriptions of study procedures. |
| Readability Software | Automatically calculates text readability scores and complexity metrics. | Provides objective, quantitative data to track improvement during the revision process [38]. |
| Plain Language Glossary | A curated list of jargon terms and their simple alternatives [37] | Serves as a direct translation guide for writers during the drafting and revision of ICFs. |
| Active Voice Style Guide | A guide promoting the use of active voice and concise sentences. | Helps restructure text to be more direct and engaging for the participant [38]. |
Core Principles of Readable Document Design
The structural design of an informed consent form is not merely an aesthetic concern; it is a fundamental component of ethical research practice. Proper formatting enhances participant comprehension, facilitates navigation, and ensures that critical information is accessible. Adherence to plain language principles is legally mandated for federal agencies by the Plain Writing Act of 2010 and represents a best practice for all research involving human subjects [39] [20]. The overarching goal is to present information in a way that the intended audience can understand it the first time they read it [20]. This involves a conscious effort to organize content to serve the audience, which includes putting the most important message first, breaking text into logical chunks, and using informative headings [20].
Structural Formatting Guidelines
Effective document structure guides the participant through the information smoothly and logically. The following protocols detail key structural elements.
Organizational Protocol
- Procedure for Information Prioritization: Begin by identifying all required elements of informed consent as per 45 CFR 46.116 [4]. Rank these elements by importance to the participant's decision-making process. The most critical information, such as the voluntary nature of research and key risks, must be presented first [20]. This often aligns with the "Key Information Elements" suggested in the revised Common Rule, which include a statement that the project is research, a summary of the research (purpose, duration, procedures), risks, benefits, and alternatives [4].
- Procedure for Chunking Text: Analyze the draft consent form and separate content into distinct, logical sections (e.g., Purpose, Procedures, Risks, Benefits, Confidentiality, Voluntary Participation). Limit each paragraph to a single topic and a maximum of five sentences [20]. Use white space to separate these sections and paragraphs, which reduces cognitive load and improves readability.
Typography and Spacing Protocol
- Procedure for Setting Readability: Aim for an 8th-grade reading level for all participant-facing materials [4] [9]. Use tools such as the Flesch-Kincaid Grade Level formula to assess reading level, keeping in mind that this formula is best for determining sentence length and other factors like sentence structure and word difficulty must be considered separately [9]. Strive for an average of 20 words per sentence and limit each sentence to one idea [20].
- Procedure for Implementing Active Voice: Review the document for sentences structured in a passive voice (e.g., "You will be asked by the researcher..."). Restructure these sentences to use an active voice (e.g., "The researcher will ask you...") [20]. This enhances clarity and directness. Consistently use the second person ("you") or third person ("he/she") to speak directly to the participant and avoid the first person ("I") in the body of the form [4].
Quantitative Data Presentation on Contrast Requirements
Visual presentation, particularly color contrast, is critical for readability. The Web Content Accessibility Guidelines (WCAG) provide measurable criteria to ensure text is perceivable by all users, including those with visual impairments [40] [41]. The following table summarizes the minimum contrast ratios for text against its background, as per WCAG.
Table 1: WCAG Color Contrast Requirements for Text Accessibility
| Text Type | Minimum (Level AA) | Enhanced (Level AAA) | Visual Example |
|---|---|---|---|
| Normal Text | 4.5:1 | 7:1 | This text is #767676 on white (#FFFFFF) |
| Large Text (18pt or 14pt bold+) | 3:1 | 4.5:1 | This text is #949494 on white (#FFFFFF) |
These requirements ensure that the visual presentation of text is accessible to users with low vision or color vision deficiencies [42] [40]. It is important to note that these ratios also apply to images of text [40]. Text that is part of a logo or is purely decorative is exempt from these requirements [40].
Experimental Protocol for Verifying Readability and Accessibility
This protocol provides a step-by-step methodology for validating the readability and visual accessibility of an informed consent form prior to IRB submission and use in a study.
Research Reagent Solutions
Table 2: Essential Tools for Readability and Accessibility Testing
| Tool Name | Type | Primary Function |
|---|---|---|
| Flesch-Kincaid Grade Level | Software Formula | Assesses the U.S. grade-level reading score of a text document [9]. |
| WebAIM Contrast Checker | Web-based Tool | Checks the contrast ratio between foreground and background hex color codes [41]. |
| Plain Language Checklist | Guideline Document | Provides a systematic list of criteria for clear and understandable language [20]. |
| WAVE Browser Extension | Browser Add-on | Identifies accessibility issues, including contrast errors, on web pages or in digital documents [41]. |
Step-by-Step Validation Procedure
The following workflow diagram outlines the experimental protocol for validating informed consent form readability and accessibility.
Procedure 1: Plain Language Application
- Gather the draft informed consent document.
- Using the Plain Language Checklist [20], systematically review the document.
- Rewrite sentences to use active voice, common words, and limit to one idea per sentence.
- Reorganize content to ensure the most important information for the participant's decision is presented first.
- Output: A revised document that adheres to plain language principles.
Procedure 2: Readability Assessment
- In Microsoft Word, access the spelling and grammar check feature, which includes a function to show the Flesch-Kincaid Reading Ease and Grade Level [9].
- Run the check on the revised document from Procedure 1.
- The target is an 8th-grade reading level or lower [4] [9]. If the score is higher, return to Procedure 1 to further simplify the language and sentence structure.
- Output: A validated readability score.
Procedure 3: Color Contrast Verification
- Identify the primary text and background color codes (e.g., HEX codes) used in the document.
- Input the foreground and background color codes into the WebAIM Contrast Checker tool [41].
- The tool will calculate the contrast ratio. Verify that all normal text meets or exceeds a 4.5:1 ratio and large text meets or exceeds a 3:1 ratio.
- If the contrast is insufficient, select new colors from the approved palette that meet the required ratios.
- Output: A document with visually accessible color choices.
Logical Framework for Document Design
The relationship between design choices and participant understanding is direct and critical. The following diagram illustrates the logical pathway from core principles to successful ethical outcomes.
The teach-back method is a evidence-based, interactive communication technique used to confirm a potential research participant's understanding of the information presented to them during the informed consent process [43]. It involves asking individuals to explain in their own words what they have just been told about the research study, including its purpose, procedures, risks, benefits, and alternatives [44]. This method transforms the consent process from a passive signing event into an active, ongoing dialogue, ensuring that consent is truly informed and voluntarily given.
Within the framework of plain language requirements for informed consent forms, teach-back serves as a critical implementation tool. It moves beyond document readability to assess and enhance actual comprehension, addressing the critical gap that often exists between providing information and confirming it is understood [45]. For researchers and drug development professionals, integrating teach-back protocols mitigates the risk of participants misunderstanding their role in a study, thereby supporting ethical standards, regulatory compliance, and data integrity.
Quantitative Evidence Supporting Teach-Back Efficacy
Robust research demonstrates the effectiveness of the teach-back method in improving health communication and adherence across various clinical contexts.
Table 1: Impact of Teach-Back on Patient Outcomes in Clinical Studies
| Study Design / Population | Key Intervention | Measured Outcomes | Results |
|---|---|---|---|
| Randomized Controlled Trial (RCT) in patients with hypothyroidism [46] | Self-care training using teach-back method over two sessions (45-60 min each) plus telephone follow-up. | Adherence to treatment (measured via validated questionnaire). | A statistically significant difference in mean overall adherence score was found between intervention and control groups (P < 0.001). Effect size (Cohen's d) was greater than 0.80. |
| Systematic Review of 26 studies (15 cohort, 5 RCTs, 5 quasi-experimental, 1 qualitative) [43] | Teach-back used to reinforce patient education in chronic diseases (e.g., heart failure, diabetes). | Disease knowledge, self-management, patient satisfaction, post-discharge readmission. | The majority of studies showed improved patient satisfaction and disease knowledge. Six studies on readmission reported statistically significant improvement or positive trends. |
| Pretest-Posttest Intervention Study in an Emergency Department setting [43] | Discharge instructions delivered using the teach-back method. | Patient knowledge of diagnosis, follow-up care, and medications. | Significantly higher knowledge scores for diagnosis (P < 0.001) and follow-up instructions (P = 0.03). Medication knowledge was higher but not statistically significant (P = 0.14). |
Experimental Protocols for Implementing Teach-Back
The following protocols provide a detailed methodology for integrating teach-back into the informed consent process for clinical research.
Core Teach-Back Discussion Protocol
This protocol outlines the fundamental steps for conducting a teach-back session during the consent discussion, before any study-related procedures begin [44].
Materials:
- Informed Consent Form (ICF) written in plain language.
- Any study-specific visual aids or diagrams.
- Access to interpreter services if required.
Procedure:
- Set the Stage: Introduce the teach-back process to the participant. Use a normal, respectful tone. Example: "I want to be sure I've explained everything clearly. So, after we go through each section, I'm going to ask you to explain it back to me in your own words. Is that okay?"
- Explain a Concept: Break down the complex study information into small, manageable chunks. Use plain language, avoiding medical jargon. For example, explain the study's primary purpose, then pause for teach-back before moving to procedures.
- Ask for a Teach-Back: Ask an open-ended, non-shaming question to check understanding.
- Instead of: "Do you understand?"
- Use: "Could you now explain back to me what we just discussed about the main reason we are doing this study?" or "Just to be sure I was clear, what would you tell your family member about the main risks of participating?"
- Assess and Clarify: Listen carefully to the participant's response.
- If the explanation is correct and complete: Acknowledge this and proceed to the next concept. Example: "Thank you, that's exactly right. Now let's talk about..."
- If the explanation is incorrect or incomplete: This indicates a need for re-teaching. Do not blame the participant. Say: "I apologize, I didn't explain that clearly enough. Let me try again." Re-explain the information using different words or methods.
- Repeat and Document: Continue this cycle of explain → ask → assess → clarify until the participant demonstrates a clear understanding of all key aspects of the study. Document in the participant's record that the teach-back method was used to confirm understanding.
Protocol for a Randomized Controlled Trial on Teach-Back Efficacy
This protocol is based on a recent clinical trial and can be adapted to study the impact of teach-back within a research population [46].
Objective: To determine the impact of self-care training using the teach-back method with telephone follow-up on adherence to treatment in patients with a chronic condition.
Study Design: Randomized Controlled Clinical Trial.
Participants:
- Inclusion Criteria: Patients (age ≥18) diagnosed with the condition for a minimum of six months, able to read and write, access to a phone, no significant cognitive or sensory impairments, willingness to participate.
- Sample Size: Determined by power analysis (e.g., 62 participants as in the referenced study).
- Randomization: Eligible participants are randomly assigned to an intervention group or a control group.
Intervention:
- Control Group: Receives routine care and standard informed consent/education.
- Intervention Group: Receives routine care plus:
- Structured Teach-Back Sessions: Two structured self-care training sessions using the teach-back method, each lasting 45-60 minutes. Key information is broken down, and participants are asked to explain concepts and instructions in their own words until proficiency is demonstrated.
- Telephone Follow-Up: Scheduled follow-up calls (e.g., at 2 weeks and 4 weeks post-education) to reinforce learning, address questions, and conduct brief remote teach-back assessments.
Data Collection:
- Tools: Validated questionnaires, such as the Adherence to Treatment Questionnaire [46], administered at baseline, immediately post-intervention, and at a follow-up time point (e.g., 2 months).
- Demographics: A demographic characteristics form is completed at baseline.
Data Analysis:
- Use statistical software (e.g., SPSS v23).
- Employ independent t-tests, Mann-Whitney tests, and Repeated Measures ANOVA to compare mean scores between groups and over time.
- A P-value of <0.05 is considered statistically significant.
Visualization of the Teach-Back Workflow
The following diagram illustrates the cyclical, non-linear process of conducting a teach-back conversation.
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Materials and Tools for Implementing Teach-Back Protocols
| Item / Tool | Function / Explanation |
|---|---|
| Plain Language Informed Consent Form (ICF) | The foundational document, written at an appropriate reading level (e.g., 6th-8th grade), using short sentences and active voice, free of complex legal and scientific jargon. This is a prerequisite for effective teach-back. |
| Validated Comprehension Assessment Questionnaires | Standardized tools to quantitatively measure participants' understanding of study key elements (e.g., purpose, risks, procedures) before and after the teach-back intervention to gauge its efficacy. |
| Structured Teach-Back Script/Guide | A protocol detailing the key concepts to be covered, along with suggested open-ended questions for each concept (e.g., "What are the two or three main things that could go wrong if you take this study drug?"). |
| Professional Interpreter Services | Certified interpreters (in-person or remote) are essential for conducting teach-back with participants whose primary language is not English, ensuring comprehension is accurately assessed and that consent is not waived [47]. |
| Digital Recording System (with consent) | To audio/video record the consent conversation (with explicit permission) for quality assurance, training purposes, and to document the interactive process for auditors or IRBs. |
| Data Collection & Analysis Software | Software such as SPSS, R, or SAS for managing participant data and performing statistical analyses (t-tests, ANOVA) to evaluate the outcomes of teach-back implementation in research settings [46]. |
Solving Common Challenges: From Complex Protocols to Vulnerable Populations
Tailoring Language for Comparative Effectiveness Research (CER) and Standard Care Trials
Comparative Effectiveness Research (CER) and standard care trials, which compare interventions already in routine clinical practice, present unique challenges for the informed consent process. Traditional, legally-oriented consent forms are often a poor fit for these studies, as they can misleadingly suggest that risks and procedures are experimental rather than part of standard care [48]. The revised U.S. Common Rule requirement for a concise key information section at the beginning of consent forms aims to address comprehension barriers, but provides little practical guidance on implementation [48] [49]. Evidence demonstrates that refining consent language to the specific context of CER can significantly improve participant understanding without adversely affecting enrollment rates [48]. This protocol provides detailed methodologies for creating, testing, and implementing plain language consent forms tailored to the ethical and practical demands of CER.
Experimental Evidence: Quantifying the Impact of Language Tailoring
Key Findings from Consent Modification Studies
Recent empirical studies have systematically evaluated the impact of tailored consent language. The following table synthesizes quantitative outcomes from key experiments, demonstrating the effects of various modifications on participant understanding and willingness to enroll.
Table 1: Impact of Consent Form Modifications on Understanding and Enrollment
| Experimental Condition | Key Modification | Effect on Understanding | Effect on Enrollment Likelihood | Citation |
|---|---|---|---|---|
| Standard vs. Tailored Injury Language | Compensation for injury language clarified to reflect standard care context. | Understanding of injury compensation process doubled (25% to 51%, p<0.0001). | No significant difference (73% vs 75%, p=0.6). | [48] |
| Modified Key Information | Key information page simplified and positively framed. | Understanding of randomization improved (from 44% to 59% in one group). | No significant difference across groups (85-88%, p=0.6). | [48] |
| Plain Language Recommendations (PLR) | Health recommendations presented in plain language vs. standard version. | Understanding significantly improved for one recommendation (MD 19.8%, 95% CI 14.7–24.9%, p<0.001). | N/A (Intention to follow recommendation increased, MD 1.2 on 7-point scale, p<0.001). | [50] |
| Comprehensive Plain Language Revision | Polish consent form rewritten using readability software and plain language principles. | Readability level reduced from university degree level to primary school (4-6 years) level. | Assessed as "completely comprehensible" by most participants. | [38] |
Detailed Experimental Protocol: Consent Language Evaluation
This protocol outlines a method for empirically testing the effectiveness of modified consent forms, based on established research designs [48] [50].
Study Design and Setting
- Design: Allocation-concealed, blinded, controlled superiority trial, complemented by qualitative analysis.
- Platform: Online survey platforms (e.g., Amazon Mechanical Turk, managed through CloudResearch) or in-person settings.
- Population: Adult volunteers, oversampling patient populations and stakeholders relevant to the CER context. Inclusion criteria should include minimum approval ratings (e.g., 98%) for online platforms to ensure response quality [48].
Interventions and Randomization
- Participants are randomly assigned in a 1:1 ratio to review one of two or more versions of a consent form for a hypothetical CER trial.
- Control Group: Receives the "Standard" consent form (Form A), typically created from an existing institutional template.
- Intervention Group(s): Receive modified forms. Examples include:
- Form B (Tailored Compensation Language): Identical to Form A except for modifications to sections on compensation for injury and insurance coverage, emphasizing that treatments are standard and care for injuries is handled like regular medical care [48].
- Form C (Modified Key Information): Contains a simplified, more positively-framed key information page.
- Form D (Clarified Costs): Identical to Form C but explicitly states no extra costs for participation [48].
Data Collection and Outcome Measures
- Primary Outcome: Hypothetical willingness to enroll, assessed via a Likert scale question (e.g., "After reading this consent form how likely are you to give permission to include your family member in this study?") and dichotomized (Unlikely/Likely) for analysis [48].
- Secondary Outcomes:
- Understanding: Measured by the proportion of correct responses to comprehension questions about study purpose, randomization, and injury compensation processes [48].
- Accessibility, Usability, and Satisfaction: Measured on 1-7 scales [50].
- Intention to Implement: Assessed via questions on likelihood to follow or share the recommendation [50].
- Quality Control: Incorporate attention-check questions within the survey to exclude inattentive respondents [48].
Sample Size and Analysis
- Sample Size Calculation: Based on the primary outcome. For example, a sample of 650 participants (325 per group) provides 80% power with a two-sided alpha of 0.05 to detect a 10% difference in enrollment likelihood [48].
- Statistical Analysis: Perform pairwise comparisons of willingness to enroll and understanding between groups using Chi-square tests. Use multiple logistic regression for adjusted analyses.
Visualization of Experimental Workflow
The following diagram illustrates the sequential workflow for designing and conducting a consent language evaluation study.
The Scientist's Toolkit: Essential Reagents and Materials
Table 2: Key Research Reagent Solutions for Consent Language Studies
| Tool/Reagent | Primary Function | Application Example | Key Features/Benefits |
|---|---|---|---|
| Readability Analysis Software (e.g., jasnopis.pl, Flesch-Kincaid) | Objectively assess the reading grade level of a consent form. | Initial benchmarking of a standard form's complexity and post-revision verification [38]. | Provides quantitative score; helps meet IRB readability requirements (often 8th grade level). |
| The RUAKI Indicator (Readability, Understandability, and Actionability of Key Information) | An 18-item validated tool to evaluate and improve the key information section of consent forms [49]. | Systematically checking a draft key information section against plain language criteria (e.g., familiar words, active voice, short chunks of text). | Specifically designed for CER consent; evidence of validity and reliability (substantial inter-rater agreement). |
| Online Participant Recruitment Platforms (e.g., Amazon Mechanical Turk via CloudResearch) | Efficiently recruit a large, diverse sample of participants for survey-based experiments. | Hosting a randomized controlled trial to compare different consent form versions [48]. | Allows for rapid data collection, quality controls (e.g., approval ratings), and broad demographic reach. |
| Plain Language Writing Guidelines | Provide principles for simplifying text (e.g., use of familiar words, active voice, short sentences and paragraphs) [49] [38]. | Rewriting a dense, legalistic "Compensation for Injury" section into clear, direct language. | Improves comprehension for individuals with varying health literacy levels; reduces patient anxiety. |
| Stakeholder Advisory Panels (Patients, clinicians, advocates) | Provide direct input on clarity, concerns, and acceptability of consent language and models. | Reviewing and refining proposed streamlined consent models (e.g., Opt-Out, General Approval) for a low-risk CER study [51]. | Ensures participant-centricity; identifies potential misunderstandings or ethical concerns early. |
Visualization of Consent Form Development and Testing Logic
The logical flow for developing and validating a tailored consent form is based on an iterative cycle of creation, testing, and refinement.
Advanced Application: Streamlined Consent Models for Learning Health Systems
Within Learning Health Systems (LHS) where CER is embedded into routine care, traditional opt-in consent can be a significant barrier [51]. Research indicates stakeholder acceptability of streamlined models depends heavily on study design and risk.
Table 3: Acceptability of Streamlined Consent Models for Different CER Studies [51]
| Consent Model | Description | Observational CER Study (Acceptability) | Randomized CER Study (Acceptability) |
|---|---|---|---|
| General Approval | Patients are notified via institutional policies/posters; no routine explanation or opt-out for specific studies. | 67% of stakeholders found this acceptable. | 11% of stakeholders found this acceptable. |
| Opt-Out | Patients receive a brief description; enrollment is automatic unless they decline. | 45% of stakeholders found this acceptable. | 54% of stakeholders found this acceptable. |
| Opt-In (Traditional) | Patients receive full written/oral information and must actively agree to participate. | 36% of stakeholders found this acceptable. | 80% of stakeholders found this acceptable. |
Protocol Note: Stakeholder deliberation sessions reveal that streamlined models (General Approval, Opt-Out) are more acceptable for observational CER studies, while traditional Opt-In consent is strongly preferred for randomized trials, even when comparing standard treatments [51]. This underscores that consent approaches must be tailored to the research context.
Quantitative Data on Injury Compensation
Compensation in personal injury cases is designed to cover the full spectrum of financial, physical, and emotional impacts experienced by the injured party. The following tables summarize key quantitative data and structural relationships essential for understanding compensation frameworks.
Table 1: Compensation Ranges by Injury Severity [52]
| Injury Severity Category | Example Injuries | Average Compensation Range for Medical Bills | Associated Compensable Damages |
|---|---|---|---|
| Minor | Cuts, bruises, soft tissue damage (whiplash) | $2,000 - $10,000 | Emergency care, diagnostic imaging, minor physical therapy [52] [53] |
| Moderate | Broken bones, concussions | $20,000 - $75,000 | Surgical procedures, extended rehabilitation, lost wages, some pain and suffering [52] [53] |
| Severe / Catastrophic | Traumatic brain injury, spinal cord damage, severe burns | Often exceeds $100,000; can reach millions | Long-term / lifetime medical care, permanent disability, full lost earning capacity, significant pain and suffering [52] |
Table 2: Breakdown of Compensable Cost Categories [52] [53] [54]
| Cost Category | Specific Examples | Classification |
|---|---|---|
| Medical Expenses | Emergency care, surgery, physical therapy, prescription medications, future medical procedures | Economic Damages |
| Financial & Tangible Losses | Lost wages, loss of future earning capacity, property damage (e.g., vehicle repair) | Economic Damages |
| Hidden & Indirect Costs | Home modifications (ramps, grab bars), specialized transportation, household services (cleaning, childcare), mental health treatment (PTSD, anxiety, depression) | Economic & Non-Economic Damages |
| Intangible Losses | Pain and suffering, emotional distress, loss of enjoyment of life, loss of consortium (relationship damage) | Non-Economic Damages |
Experimental Protocols for Compensation Analysis
Protocol for Documenting and Calculating Comprehensive Damages
Objective: To establish a standardized methodology for quantifying the full financial impact of a research-related injury, ensuring all economic and non-economic damages are accurately assessed for inclusion in informed consent communications and potential compensation.
Workflow Diagram: Compensation Calculation Workflow
Materials:
- Financial Documentation Portfolio: A comprehensive system for collecting all injury-related receipts, bills, and proof of payment [53].
- Medical Expert Testimony: Professional assessments from treating physicians and specialists to project long-term treatment needs and permanent disability [54].
- Vocational & Economic Expert Analysis: Reports to quantify loss of earning capacity and future wage loss, supported by employer statements [53] [54].
- Legal Calculation Frameworks: Validated methodologies, such as the Multiplier and Per Diem methods, for quantifying non-economic damages like pain and suffering [55].
Procedure:
- Documentation Phase: Collect every expense related to the injury from the date of incident onward. This includes, but is not limited to, medical bills, pharmacy receipts, transportation costs for medical appointments, invoices for home modifications, and receipts for hired household help. Simultaneously, document lost work hours and corresponding lost wages [53].
- Categorization Phase: Separate all documented costs into economic and non-economic damages as defined in Table 2.
- Economic Damages Quantification:
- Sum all past and current medical expenses and verified lost income.
- Work with medical and economic experts to calculate the present value of future medical costs and loss of future earning capacity [54].
- Non-Economic Damages Quantification: Apply one of two standard legal methods to calculate compensation for pain, suffering, and emotional distress [55]:
- Multiplier Method: Total the economic damages and multiply by a factor (typically 1.5 to 5), with the severity of the injury justifying a higher multiplier.
- Per Diem Method: Assign a reasonable daily dollar value to the victim's pain and suffering, then multiply by the number of days of recovery.
- Final Calculation: Sum the totals from quantified economic damages (including future costs) and non-economic damages to arrive at a comprehensive compensation figure.
Protocol for Analyzing Insurance Policy Limitations
Objective: To systematically identify and evaluate the available insurance coverage and its limits, which often dictate the maximum potential recovery in an injury claim, and to explore strategies for addressing coverage shortfalls.
Workflow Diagram: Insurance Limitation Analysis
Materials:
- Insurance Policy Disclosures: The relevant liability insurance policies (e.g., auto, homeowner's, business) from the at-fault party [56].
- Personal Insurance Policy Documentation: The injured party's own insurance policies, specifically examining underinsured (UIM) and uninsured (UM) motorist coverage provisions [56].
- Asset Investigation Tools: Legal means to identify other potentially liable parties and the personal assets of the at-fault party.
Procedure:
- Policy Identification: Obtain the liability insurance policy of the at-fault individual or entity.
- Limit Assessment: Determine the specific policy limits, which are typically stated as a maximum "per person" and "per accident" amount. Note that these limits are the maximum the insurer is obligated to pay, regardless of the total damages incurred [56].
- Gap Analysis: Compare the total calculated damages (from Protocol 2.1) against the available policy limits to identify any shortfall.
- Alternative Avenues Exploration: If a shortfall exists, investigate all potential sources for additional compensation [56]:
- UIM/UM Claims: File a claim against the injured party's own underinsured or uninsured motorist coverage, if available.
- Multiple Defendants: Identify any other legally responsible parties (e.g., employers, manufacturers, property owners) whose insurance policies may be accessed.
- Asset Recovery: In cases of severe negligence, investigate the possibility of recovering compensation directly from the at-fault party's personal assets or any umbrella liability policies they may hold.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Materials for Injury Cost Documentation and Analysis
| Item | Function |
|---|---|
| Financial Documentation Portfolio | A centralized system (digital or physical) for storing every receipt, bill, and proof of payment related to the injury, forming the foundational evidence for economic damages [53]. |
| Medical & Vocational Expert Assessments | Professional reports from doctors and vocational specialists that objectively validate long-term medical needs, permanent impairment, and loss of earning capacity, which are critical for justifying future costs [54]. |
| Legal Calculation Frameworks (Multiplier/Per Diem) | Standardized, court-accepted methodologies for quantitatively translating intangible harms like pain and suffering into a monetary value, ensuring compensation demands are legally and logically sound [55]. |
| Comprehensive Insurance Policy Review | A thorough analysis of all relevant insurance policies (at-fault party and personal) to identify available coverage, critical limits, and potential barriers to full financial recovery [56]. |
| Journal of Pain & Lifestyle Impact | A daily log maintained by the injured party documenting their pain levels, emotional state, and specific activities they can no longer perform, providing subjective evidence to support claims for non-economic damages [54]. |
Informed consent is a fundamental ethical requirement in human subjects research, representing more than just a signed form—it is a comprehensive process that ensures participants voluntarily confirm their willingness to participate after understanding all relevant aspects of the research [57]. The plain language principle transforms this process from a legal formality into a meaningful dialogue. The Minnesota Department of Health IRB defines plain language as communication your audience can understand the first time they read or hear it, enabling them to find what they need, understand what they find, and use what they find to meet their needs [58].
For vulnerable populations—including children, cognitively impaired individuals, and the critically ill—the adaptation of consent processes using plain language becomes particularly crucial. These groups face unique challenges in understanding complex research information and making autonomous decisions. Providing clear communication is one of the key ways researchers can foster trust, understanding, and dialogue with potential participants and their communities [58]. When consent materials are not presented in plain language, they fail to achieve their intended purpose of truly informing participants, thereby undermining the ethical foundation of the research [58].
Plain Language Foundations and Regulatory Framework
Core Principles of Plain Language
The application of plain language in informed consent is supported by several evidence-based principles. These principles are not about "dumbing down" information but rather about communicating complex information clearly and effectively [58]. Key principles include:
- Logical organization: Presenting information in an order that makes sense to the reader
- Single-theme paragraphs: Limiting each paragraph to one main idea
- Clear headings and subheadings: Breaking information into manageable sections
- Bulleted lists: Using visual cues to present related items
- Active voice and concrete nouns: Creating more engaging and understandable text
- Conversational style: Using pronouns like "you" and "we" to create connection
- Jargon elimination: Removing or explaining technical and scientific terms
The Revised Common Rule mandates that consent forms "begin with a concise and focused presentation of the key information that is most likely to assist a prospective subject or legally authorized representative in understanding the reasons why one might or might not want to participate in the research" [2]. This regulatory requirement emphasizes the importance of presenting the most critical information clearly at the beginning of the consent document.
Readability Assessment and Documentation
For materials intended for the general adult population, best practices in health literacy recommend aiming for a sixth-grade reading level or lower [58]. The Flesch-Kincaid readability tool in Microsoft Word can assess reading level, but researchers should be aware that readability software has limitations—it doesn't account for organization, formatting, or conceptual complexity [58].
Table 1: Plain Language Checklist for Informed Consent Documents
| Category | Checkpoint | Compliance |
|---|---|---|
| Organization | Information presented in logical order | ☐ |
| Adequate white space and margins | ☐ | |
| Each paragraph has single theme | ☐ | |
| Language | Jargon and technical terms eliminated | ☐ |
| Active voice and concrete nouns used | ☐ | |
| Short words and sentences | ☐ | |
| Design | Clear, meaningful headings | ☐ |
| Appropriate bulleted/numbered lists | ☐ | |
| Readable font size and type | ☐ | |
| Content | Anticipates reader questions | ☐ |
| Necessary technical terms explained | ☐ | |
| Age- and culturally-appropriate | ☐ |
Pediatric Consent and Assent Protocols
Conceptual Framework: Consent, Assent, and Dissent
In pediatric research, the concept of consent is complex and involves multiple components. Parental permission serves as the formal consent for a child to participate in research, while assent represents the child's affirmative agreement to participate [59]. The underlying ethical value differs between adults and children—for adults, the primary justification is autonomy, while for children, it is respect for persons, focusing on the welfare and interests of the child [59].
A key feature of child assent is that it does not need to be burdened with the same informational and decision-making standards as adult informed consent [59]. Assent provides children with the opportunity to choose to the extent that they are able, giving recognition to the value of the role for children that lies between no involvement in discussions and full decisional authority [59].
Developmental Approach to Pediatric Assent
The approach to assent should be developmentally appropriate, recognizing that children's capacities evolve as they mature. The assent process is sometimes attributed value in promoting children's moral growth and developing autonomy, allowing them to obtain practice in making decisions and contributing to their perception that they have some control over their lives [59].
Table 2: Developmental Considerations for Pediatric Assent
| Age Group | Capacity Level | Assent Approach |
|---|---|---|
| 0-6 years | Limited understanding | Focus on comfort, use play and demonstration |
| 7-11 years | Emerging understanding | Simple explanations, check for basic comprehension |
| 12-14 years | Growing capacity | More detailed information, respect emerging autonomy |
| 15-17 years | Near-adult capacity | Similar to adult consent with appropriate support |
Dissent, defined as a child's objection or expression of disapproval, carries substantial weight and requires a lower level of understanding than assent [59]. Even children who are too immature to provide formal assent may be able to register dissent. For these children, overriding dissent is justified only in exceptional circumstances, such as when an intervention offers direct benefit that is important to the child's health or well-being and is available only in the context of the research [59].
Experimental Protocol: Implementing Pediatric Assent
Protocol Title: Developmentally Appropriate Assent Process for Pediatric Research Participants
Purpose: To obtain meaningful assent from child participants while maintaining ethical standards and regulatory compliance.
Materials Needed:
- Age-appropriate assent documents
- Visual aids and demonstration materials
- Private, comfortable environment
- Teach-back explanation materials
- Parental permission documentation
Procedure:
- Pre-assessment Phase
- Determine child's developmental stage and cognitive capacity
- Prepare age-appropriate materials (simplified forms, visual aids)
- Ensure parental presence based on child's preference and situation
Information Disclosure Phase
- Explain research procedures using concrete, relatable terms
- Use developmentally appropriate methods (play, diagrams, stories)
- Clearly state voluntary nature of participation
- Explain that saying "no" will not result in punishment or disappointment
Comprehension Assessment Phase
- Ask child to explain the research in their own words
- Use teach-back method to verify understanding
- Encourage questions and provide clear answers
- Assess both verbal and non-verbal cues
Decision Phase
- Explicitly ask for the child's agreement to participate
- Respect and document dissent without coercion
- Provide ongoing opportunities to withdraw assent
- Document the assent process thoroughly
Validation Measures:
- Use age-appropriate comprehension assessment tools
- Document child's questions and responses
- Include independent assent monitor for higher-risk studies
- Implement ongoing assent verification throughout study duration
Consent Protocols for Cognitively Impaired Individuals
Capacity Assessment and Surrogate Decision-Making
For cognitively impaired individuals, the initial step involves assessing decision-making capacity. As a general rule, all adults, regardless of diagnosis or condition, should be presumed competent to consent unless there is evidence of a serious mental disability that would impair reasoning or judgment [60]. Cognitive impairment alone should not automatically disqualify a person from consenting to research participation; rather, there should be specific evidence of individuals' incapacity to understand and make a choice before they are deemed unable to consent [60].
When individuals lack capacity, consent may be sought from a legally authorized representative. The hierarchy of surrogate decision-makers typically includes: the patient's spouse; adult children; parents; or someone previously identified by the patient before becoming incapacitated [60]. Surrogate decision-makers should base their decisions on knowledge of what the patient would desire, if known.
Risk Classification and Safeguards
Research involving cognitively impaired persons requires additional safeguards, particularly regarding risk levels. Research that presents more than minimal risk should involve cognitively impaired persons only when the research holds prospects of direct benefit to these individuals [60]. A minor increase over minimal risk may be permitted in research involving institutionalized individuals only where research is designed to evaluate an intervention of foreseeable benefit to their care.
Table 3: Risk-Benefit Framework for Research with Cognitively Impaired Persons
| Risk Level | Direct Benefit | Allowable Research | Additional Safeguards |
|---|---|---|---|
| Minimal risk | Not required | Research on condition or circumstances | Regular capacity reassessment |
| Minor increase over minimal risk | Possible but not required | Research related to condition | Independent consent monitor |
| More than minimal risk | Required | Research with direct therapeutic benefit | Surrogate consent + participant assent |
Essential safeguards for research with cognitively impaired individuals include:
- Independent assessment of capacity by qualified professionals
- Documentation of capacity assessment with formal dated and signed notes
- Solicitation of participant assent to the extent possible
- Ongoing monitoring of changing capacity throughout research
- Independent advocacy for participants when appropriate
Experimental Protocol: Capacity Assessment and Consent
Protocol Title: Capacity Assessment and Adaptive Consent Process for Cognitively Impaired Adults
Purpose: To ethically enroll cognitively impaired individuals in research through appropriate capacity assessment and surrogate decision-making when necessary.
Materials Needed:
- Validated capacity assessment tools
- Documentation forms for capacity assessment
- Consent forms for legally authorized representatives
- Simplified information materials for participants
- Independent monitor documentation forms
Procedure:
- Initial Capacity Screening
- Conduct preliminary assessment using standardized tools
- Evaluate understanding, appreciation, reasoning, and expression of choice
- Document findings with specific examples of demonstrated capacity or incapacity
- Include input from treating physicians when appropriate
Adaptive Consent Process
- For capable individuals: proceed with standard informed consent process
- For questionably capable: implement enhanced consent process with simpler language, repetition, and comprehension checks
- For incapable individuals: identify legally authorized representative according to state hierarchy
Surrogate Consent Implementation
- Verify representative's authority according to applicable law
- Ensure representative understands their role in deciding based on patient's values
- Provide complete research information to surrogate
- Document surrogate decision-making process
Participant Involvement
- Solicit assent from incapable individuals using simplified explanations
- Respect dissent and document objections
- Provide ongoing information appropriate to comprehension level
- Reassess capacity periodically for fluctuating conditions
Validation Measures:
- Use standardized capacity assessment instruments (MacCAT-CR, UBACC)
- Document comprehension using teach-back method
- Include independent consent monitor for higher-risk studies
- Implement periodic capacity reassessment for progressive conditions
Emergency and Critical Care Consent Protocols
Exception Circumstances and Consent Waivers
In emergency and critical care research, obtaining prospective informed consent may not always be possible or practical. The Common Rule permits waiver of informed consent under specific circumstances when: (1) the research involves no more than minimal risk; (2) the waiver will not adversely affect participants' rights and welfare; (3) the research could not practicably be carried out without the waiver; and (4) participants will be provided with additional pertinent information after participation when appropriate [57].
For emergency research, specific conditions must be met to justify waived or deferred consent, including: subjects affected by a life-threatening condition; the investigational treatment is experimental; it is impracticable to obtain consent; and the waiver of informed consent is needed for the clinical trial [57]. In these circumstances, researchers must attempt to contact the legally authorized representative, and family members should have the opportunity to decline participation on behalf of the patient.
Risk-Benefit Evaluation in Critical Care Research
Ethical research in critical care settings requires careful evaluation of risks and benefits, particularly distinguishing between therapeutic procedures (with potential direct benefit to participants) and non-therapeutic procedures (performed solely to answer research questions) [61]. The standard of clinical equipoise must be met for therapeutic procedures, meaning genuine uncertainty exists within the expert medical community about the preferred treatment [61].
Experimental Protocol: Emergency Research Consent
Protocol Title: Deferred Consent and Community Consultation Protocol for Emergency Research
Purpose: To ethically conduct research in emergency settings where prospective consent is not feasible while maintaining respect for participants and communities.
Materials Needed:
- Community consultation documentation forms
- Deferred consent information materials
- Regulatory approval documentation
- Surrogate contact protocols
- Public disclosure materials
Procedure:
- Pre-Study Community Consultation
- Conduct community meetings to discuss research purpose and methods
- Solicit feedback on research acceptability and consent approach
- Document community consultation process and responses
- Modify research plan based on community input when feasible
Regulatory Approval Process
- Submit research protocol to IRB with justification for consent waiver
- Demonstrate compliance with regulatory requirements for exception
- Provide plan for deferred consent and ongoing information disclosure
- Obtain appropriate regulatory approvals before study initiation
Participant Enrollment Process
- Identify eligible participants according to study criteria
- Attempt to locate legally authorized representative when possible
- Document attempts to obtain consent and reasons for impracticability
- Enroll participants according to approved protocol
Deferred Consent and Information Disclosure
- Approach participants or surrogates as soon as feasible after enrollment
- Provide complete information about research participation
- Obtain consent for continued participation and use of data
- Offer opportunity to withdraw without penalty
Validation Measures:
- Document community consultation process and outcomes
- Record attempts to contact surrogates and obtain consent
- Track participant/surrogate understanding during deferred consent
- Monitor continued willingness to participate after full disclosure
Research Reagent Solutions Toolkit
Table 4: Essential Research Reagents and Materials for Consent Adaptation Research
| Reagent/Material | Function/Application | Implementation Example |
|---|---|---|
| Readability Software | Assess reading level of consent materials | Flesch-Kincaid tool in Microsoft Word to achieve ≤8th grade level [58] |
| Plain Language Guidelines | Create understandable consent documents | CDC's Everyday Words for Public Health Communication [58] |
| Capacity Assessment Tools | Evaluate decision-making capacity | MacCAT-CR, UBACC for cognitively impaired individuals [60] |
| Developmental Assessments | Determine appropriate assent approach | Piaget-based cognitive staging for pediatric participants [59] |
| Teach-Back Materials | Verify comprehension of consent information | Simplified visual aids and question guides for all populations [2] |
| Cultural Adaptation Frameworks | Ensure cultural appropriateness | Guidelines for translation and localization for diverse populations [58] |
Adapting informed consent for vulnerable populations requires both ethical commitment and methodological rigor. The application of plain language principles is not merely a regulatory requirement but a fundamental component of ethical research practice. For children, this means implementing developmentally appropriate assent processes that recognize their evolving autonomy. For cognitively impaired individuals, it necessitates careful capacity assessment and meaningful involvement to the extent possible. For critically ill patients, it may require innovative approaches like deferred consent when prospective consent is not feasible.
Successful implementation requires ongoing assessment of comprehension, cultural sensitivity, and respect for the unique vulnerabilities and strengths of each population. By adopting these evidence-based protocols and approaches, researchers can ensure that the consent process truly respects persons and upholds the highest ethical standards in research with vulnerable populations.
The management of multi-site and international clinical trials is a complex endeavor, characterized by significant institutional and regional variations that can impact study integrity, timelines, and compliance. The recent adoption of updated international guidelines, including ICH E6(R3) for Good Clinical Practice and the EU Clinical Trials Regulation, has substantially altered the regulatory landscape for trial sponsors and Contract Research Organizations (CROs) [62] [63]. These changes coincide with evolving ethical expectations, particularly regarding transparency and patient-centricity, which now mandate the provision of plain language summaries (PLS) of trial results to participants [64].
This application note examines the operational, regulatory, and ethical challenges inherent in multi-jurisdictional trial management. It provides detailed protocols for navigating institutional variations while maintaining compliance with the latest global standards, with particular emphasis on implementing plain language summaries within the informed consent process and results dissemination.
Regulatory Framework and Key Challenges
Current Regulatory Landscape
The table below summarizes the pivotal 2025 regulatory updates affecting multi-site and international trials.
Table 1: Key 2025 Global Regulatory Updates Impacting Multi-Site and International Trials
| Regulatory Body/Guideline | Key Update | Effective Timeline | Primary Impact on Multi-Site Trials |
|---|---|---|---|
| ICH E6(R3) [62] [63] | Principle-based GCP framework emphasizing quality by design (QbD) and risk-proportionate management. | Adopted January 2025 | Replaces prescriptive checklist approach; requires critical thinking for site-specific processes and evidence-based oversight. |
| EU Clinical Trials Regulation (CTR) [63] | Full transition to the Clinical Trials Information System (CTIS) for all applications and management. | Mandatory from January 31, 2025 | Unified submission for all concerned EU Member States via CTIS, requiring harmonized documentation and management of country-specific timelines. |
| FDA Guidance [63] | Expanded guidance on Decentralized Clinical Trials (DCTs), incorporating digital health technologies and remote activities. | 2025 | Emphasizes oversight, data integrity, and participant safety for remote and hybrid trial components across sites. |
| Various (Ethical Standard) | Updated Declaration of Helsinki (2024 revision) [64]. | 2024 | Strengthens ethical standards for transparency and participant access to information, supporting PLS initiatives. |
Implementation Challenges for Plain Language Summaries
Despite clear regulatory and ethical mandates, sponsors face significant hurdles in scaling plain language summary implementation across diverse sites and regions [64].
- Legal and Regulatory Hesitation: Legal teams often express caution regarding liability, even though no regulations prohibit the posting of PLS. This concern stems from uncertainty about promotional guidelines rather than actual regulatory barriers [64].
- Informed Consent Complications: A common barrier, especially for legacy studies, is the perception that sharing results is not permitted if PLS were not explicitly mentioned in the original informed consent forms [64].
- Site Engagement and Operational Reality: Clinical sites frequently deprioritize transparency activities due to resource constraints. This creates a disconnect, where even well-developed PLS may never reach the intended participants [64].
- Cross-Departmental Communication Gaps: Internal misalignment between clinical operations, regulatory, and disclosure teams can stall PLS initiatives, as responsibility for new GCP compliance requirements may be unclear [64].
Experimental Protocols and Methodologies
This protocol provides a step-by-step methodology for integrating PLS into multi-site international trials, from consent through dissemination.
Title: PLS Implementation Workflow
Phase 1: Pre-Trial Planning
- Action 1.1: Draft and secure cross-functional approval for standardized consent form language that explicitly mentions the future availability of a PLS. Sample language: “When study results become available, we will make a plain language summary accessible to you that explains the findings in nontechnical terms. You can find information about accessing these results at [study-specific link or general platform reference].” [64]
- Action 1.2: Define the PLS workflow in the trial protocol, specifying roles and responsibilities for writing, reviewing, approving, and disseminating the PLS. Align this with the quality by design (QbD) principles of ICH E6(R3) [63].
Phase 2: Trial Initiation
- Action 2.1: Finalize the study-specific PLS template, ensuring it is culturally and linguistically adapted for all participating regions.
- Action 2.2: Proactively engage legal and regulatory teams to mitigate perceived risks. Emphasize that PLS represent a translation of already-disclosed information (e.g., from ClinicalTrials.gov) and not new disclosure risk [64].
Phase 3: Trial Conduct and Close-Out
- Action 3.1: Execute participant recruitment using the updated informed consent forms.
- Action 3.2: Upon database lock, analyze results and draft the PLS using the pre-approved template. The writing process should involve medical writers with patient-facing communication expertise.
Phase 4: Post-Trial Dissemination
- Action 4.1: Disseminate the finalized PLS through chosen platforms. For global accessibility, consider sponsor-neutral platforms like TrialSummaries.com, which can reach 165+ countries and reduce the burden on individual sites [64].
- Action 4.2: Actively notify participants and sites of the PLS availability, fulfilling the ethical and regulatory commitment [64].
Protocol for Navigating Regional Regulatory Variations
This protocol outlines a systematic approach to managing submissions and conduct across different regulatory jurisdictions.
Step 1: Establish a Centralized Regulatory Intelligence Function
- Create a dedicated team or designate personnel within the CRO or sponsor to continuously monitor and interpret regulatory updates from key agencies (e.g., FDA, EMA, Health Canada, NMPA) [63]. This team maintains a living "Regulatory Intelligence Database."
Step 2: Conduct a Gap Analysis for Core Submission Dossier
- Develop a core, master submission dossier aligned with the most stringent regulatory requirements among the target regions. For each additional country, conduct a gap analysis to identify required country-specific amendments, documents, or formats [63].
Step 3: Implement a Unified Submission Management System
- For trials in the European Union, all submissions must be managed exclusively through the Clinical Trials Information System (CTIS) [63]. For global trials, implement a project management system that tracks disparate national timelines, review cycles, and response deadlines from a single dashboard.
Step 4: Standardize Vendor and Site Oversight with Regional Flexibility
- Develop a centralized, risk-based vendor qualification and monitoring system. While the core standards (e.g., data integrity, GCP) are universal, oversight plans should be flexible enough to accommodate regional variations in site infrastructure and standard clinical practice [63].
The Scientist's Toolkit: Research Reagent Solutions
The table below details essential materials and solutions for managing modern multi-site trials.
Table 2: Essential "Research Reagent Solutions" for Multi-Site and International Trials
| Item/Solution | Function/Description | Application in Trial Management |
|---|---|---|
| Electronic Data Capture (EDC) System | Software platform for collecting clinical data electronically in a standardized format. | Replaces paper Case Report Forms (pCRFs), improving data quality, completeness, and speeding database lock. Essential for remote data verification in risk-based monitoring [65]. |
| Clinical Trials Information System (CTIS) | The single-entry point for submission and supervision of clinical trials in the EU. | Mandatory for managing the unified application process, regulatory communication, and public disclosure for all clinical trials in the European Union [63]. |
| Plain Language Summary (PLS) Platform | A centralized, often sponsor-neutral, web platform for hosting and disseminating trial results in lay language. | Addresses key operational barriers to PLS dissemination by providing a global, consistent access point for participants, reducing legal concerns and site burden [64]. |
| Risk-Based Monitoring (RBM) Tools | Analytical software that uses statistical methods to identify atypical data patterns and potential risks across sites. | Central to the ICH E6(R3) framework; enables targeted, on-site monitoring focused on critical data and processes, improving oversight efficiency [63]. |
| Trial Master File (eTMF) System | An electronic system for the storage, management, and tracking of essential trial documents. | Ensures real-time access to compliance documents for sponsors and sites globally, which is critical for audit readiness and managing regional documentation requirements. |
Data Presentation and Analysis
Quantitative Analysis of Data Collection Methods
Empirical evidence demonstrates the operational advantages of electronic data capture systems, which are foundational for efficient multi-site management.
Table 3: Comparative Analysis of Electronic vs. Paper Case Report Forms (CRFs)
| Metric | Electronic CRFs (eCRFs) | Paper CRFs (pCRFs) | Significance/Notes |
|---|---|---|---|
| Total Cost per Patient | 374€ ±351 [65] | 1,135€ ±1,234 [65] | eCRFs showed significantly lower and less variable cost per patient. |
| Time to Database Lock | 31.7 months (Q1=24.6; Q3=42.8) [65] | 39.8 months (Q1=31.7; Q3=52.2) [65] | Trend towards shorter duration with eCRFs (p=0.11). |
| Stakeholder Preference | 31/72 respondents [65] | 15/72 respondents [65] | eCRFs were globally preferred for easier monitoring and improved data quality. |
| Typical Use Case | Large, multicenter, national Phase 3 trials with high patient numbers [65]. | Trials with fewer patients and centers [65]. | The number of patients was the only explanatory variable for CRF choice. |
Successfully managing multi-site and international trials in the current regulatory environment demands a proactive, strategic, and integrated approach. The adoption of ICH E6(R3) and the full implementation of the EU CTR necessitate a shift towards principle-based, risk-proportionate oversight [62] [63]. Furthermore, the ethical imperative of patient-centricity is now a regulatory expectation, concretized by the requirement to provide trial results in plain language [64].
As demonstrated, operational excellence can be achieved by leveraging electronic data capture systems, centralized regulatory intelligence, and standardized yet flexible processes for informed consent and results dissemination. By systematically addressing institutional and regional variations through the detailed protocols and toolkits provided, sponsors and CROs can enhance compliance, improve efficiency, and, most importantly, honor the contribution of trial participants through transparent communication.
Informed Consent Form (ICF) development represents a critical yet resource-intensive process in clinical research, consuming up to 12% of a clinical trial's budget during study conduct [66]. This document establishes application notes and detailed protocols for streamlining ICF workflows, framed within broader thesis research on plain language requirements. Optimization strategies target the entire ICF lifecycle—from initial creation using AI-driven content assembly through rigorous review and robust version control systems that prevent compliance risks [67] [66]. Implementing these structured approaches ensures regulatory compliance, enhances patient comprehension, and yields significant efficiency gains in drug development timelines.
The following sections provide a comprehensive framework for researchers and clinical operations professionals, integrating quantitative efficiency data, step-by-step experimental protocols, and visualization of optimized workflows. These evidence-based strategies address fundamental challenges in ICF management, including manual processes that increase error risk, version control failures that trigger audit findings, and localization delays that impact study startup [67] [66].
Quantitative Analysis of ICF Workflow Challenges
Resource allocation and common compliance issues in ICF workflows present significant operational hurdles. The following data summarizes key challenges and their impacts:
Table 1: Resource Allocation and Compliance Challenges in ICF Workflows
| Challenge Category | Specific Issue | Quantitative Impact | Frequency in Clinical Trials |
|---|---|---|---|
| Resource Allocation | Budget consumption during study conduct | Up to 12% of total clinical trial budget [66] | All trials |
| Manual workflow burden | Significant time investment in authoring, review, feedback incorporation [66] | Majority of organizations | |
| Version Control | Use of outdated ICF versions | Subject cohorts deemed improperly consented [67] | Common finding in audits [67] |
| Poor version labeling | Inability to identify correct form used, compromising data integrity [67] | Frequent site issue | |
| Localization & Compliance | Multiple country/customization requirements | Dozens of document versions per multi-country trial [66] | All global trials |
| Ethics committee review cycles | Extended study startup timelines [66] | All trials requiring ethics approval |
Optimized ICF Workflow Protocol
Workflow Visualization
The following diagram maps the optimized, multi-stage workflow for ICF creation, review, and version control, integrating AI efficiency and rigorous quality checks:
Stage 1: AI-Assisted ICF Creation
Objective: Leverage artificial intelligence and structured content management to accelerate initial ICF development while ensuring compliance with plain language requirements and regulatory standards.
Experimental Protocol:
Step 1: Content Modularization
- Implement a Component Content Management System (CCMS) to break ICFs into reusable components (e.g., standard risk statements, procedure descriptions, eligibility criteria) [66].
- Tag each component with metadata including protocol ID, plain language score, and regulatory requirement status.
Step 2: AI-Driven Assembly
Step 3: Plain Language Optimization
- Apply automated readability scoring (targeting 8th grade level) with highlighting of complex terminology [4].
- Implement AI-powered simplification suggestions for technical medical terminology while preserving scientific accuracy.
Quality Control Measures:
- Validate AI output against protocol requirements checklist.
- Verify plain language compliance using Flesch-Kincaid Grade Level assessment.
- Conduct technical accuracy review by medical writer.
Stage 2: Multi-Phase Review Process
Objective: Execute efficient, comprehensive review cycle incorporating stakeholder feedback while maintaining document control.
Experimental Protocol:
Step 1: Cross-Functional Review
- Distribute AI-generated draft to medical, regulatory, legal, and clinical operations stakeholders through structured content platform with role-based permissions [66].
- Implement sequential review workflow with 72-hour turnaround expectations for each stakeholder group.
Step 2: Plain Language Validation
Step 3: Ethics Committee Alignment
- Pre-submission consultation with key ethics committee members for complex studies.
- Incorporate feedback directly into CCMS for continuous model training and future improvement [66].
Quality Control Measures:
- Maintain audit trail of all comments and revisions in content platform.
- Document plain language validation methodology for regulatory inspection.
- Final sign-off from all functional leads before IRB submission.
Stage 3: Version Control Implementation
Objective: Establish robust version control system to prevent use of outdated ICFs and ensure audit-ready compliance.
Experimental Protocol:
Step 1: Version Control Log Maintenance
- Create dedicated ICF version control log tracking: version number, version date, IRB approval date, implementation date, and changes made [67].
- Restrict log editing permissions to designated study manager with read-only access for all other study team members.
Step 2: Archival Procedure Implementation
- Physically archive obsolete ICF versions in separate binder tab clearly marked "OBSOLETE" [67].
- Implement electronic equivalent in trial master file (TMF) with appropriate document superseding relationships.
Step 3: Point-of-Consent Verification
- Establish process for confirming correct ICF version immediately before each consent conversation [67].
- Cross-reference ICF footer version/date with IRB approval letter and version control log at every subject screening visit.
Quality Control Measures:
- Conduct periodic (monthly) version control audits comparing TMF documentation with site utilization records.
- Include version control verification in monitoring visit checklists with 100% source document verification.
- Document staff training on new ICF versions with signed training logs.
Research Reagent Solutions for ICF Workflow Optimization
Table 2: Essential Tools and Systems for Efficient ICF Management
| Tool Category | Specific Solution | Function in Workflow |
|---|---|---|
| Content Management | Component Content Management System (CCMS) | Stores modular ICF components (risk statements, procedures) for reusable, consistent assembly [66] |
| AI & Automation | Natural Language Processing (NLP) Tools | Identifies duplicate content, populates ICF drafts from protocols, ensures plain language compliance [66] |
| Quality Control | Readability Assessment Software | Scores text at 8th grade level, highlights complex terms for simplification to meet ethical guidelines [4] |
| Document Control | Version Control Log (Electronic or Paper) | Tracks ICF versions, approval/implementation dates, changes; prevents outdated form use [67] |
| Training & Compliance | Electronic Signature Systems | Documents staff training on new ICF versions; provides audit trail for version control procedures [67] |
Implementing these structured protocols for ICF creation, review, and version control transforms a traditionally burdensome process into an efficient, compliant, and patient-centered workflow. Integration of AI-driven content assembly with rigorous plain language validation addresses both operational efficiency and ethical imperatives for participant comprehension. The critical version control procedures mitigate significant compliance risks that can compromise study validity [67]. This comprehensive approach—combining technological innovation with systematic quality control—provides researchers and drug development professionals with a validated framework for optimizing ICF management while advancing the science of informed consent through plain language research.
Measuring Impact: Evidence and Methods for Validating Consent Improvements
Within the context of clinical research and drug development, the principle of informed consent is a cornerstone of ethical practice. The fundamental goal is to ensure that potential research participants can adequately understand the information presented to them, thereby enabling autonomous decision-making. This document frames the application of readability metrics, specifically the Flesch-Kincaid tests, within a broader thesis on plain language requirements for informed consent forms. It provides detailed protocols for researchers and scientists to quantitatively assess and improve their consent documents, aiming for the widely recommended 8th-grade reading level to enhance participant comprehension and meet regulatory expectations [4] [9].
Understanding Flesch-Kincaid Readability Tests
The Flesch-Kincaid readability tests are established tools designed to objectively measure how difficult a passage in English is to understand. They were developed to provide a quantitative measure of readability, moving beyond subjective assessment.
The Two Primary Scores
The tests produce two distinct scores, both based on the core measures of sentence length and word length, but with different weighting factors [68].
- Flesch Reading Ease (FRE): Developed by Rudolf Flesch in the 1940s, this score rates text on a scale from 0 to 100 [69] [70]. A higher score indicates that the text is easier to read.
- Flesch-Kincaid Grade Level (FKGL): Developed later for the U.S. Navy by J. Peter Kincaid and his team, this score translates readability into a U.S. grade level [71] [68]. A score of 8.0, for example, means that an eighth grader should be able to understand the text.
The Mathematical Formulas
The formulas for these scores are calculated as follows:
- Flesch Reading Ease Score:
206.835 - 1.015 * (Total Words / Total Sentences) - 84.6 * (Total Syllables / Total Words)[69] [68] [72] - Flesch-Kincaid Grade Level:
0.39 * (Total Words / Total Sentences) + 11.8 * (Total Syllables / Total Words) - 15.59[69] [71] [68]
These formulas show that both scores are sensitive to average sentence length (Total Words/Total Sentences) and average word complexity (Total Syllables/Total Words), though they weigh these factors differently.
Interpreting Scores for Informed Consent
For informed consent forms to be accessible to a broad population, including those with varying health literacy levels, a specific readability target is recommended. The following tables provide a detailed interpretation of the scores.
Table 1: Interpreting the Flesch Reading Ease Score
| Score Range | Readability Level | Typical Text Examples | Recommendation for Consent |
|---|---|---|---|
| 90-100 | 5th Grade | Very easy to read, children's literature | Too simple for complex research information |
| 80-90 | 6th Grade | Easy to read, conversational English | Good for key information summaries |
| 70-80 | 7th Grade | Fairly easy to read | Good target for informed consent forms [70] |
| 60-70 | 8th & 9th Grade | Plain English | Primary target for informed consent forms [4] [9] |
| 50-60 | 10th to 12th Grade | Fairly difficult to read | Too complex for general consent forms |
| 30-50 | College | Difficult to read | Avoid for participant-facing documents |
| 0-30 | College Graduate | Very difficult to read | Inappropriate for consent forms |
Table 2: Interpreting the Flesch-Kincaid Grade Level Score
| Flesch-Kincaid Score | Corresponding US Educational Level | Typical Usage | Recommendation for Consent |
|---|---|---|---|
| 1.0 - 6.0 | Elementary School | Children's books, beginner materials | Too simplistic for most research details |
| 6.0 - 9.0 | Middle School | Most adult fiction, newspapers | Ideal range for informed consent forms [70] [73] |
| 10.0 - 12.0 | High School | Academic texts, some technical writing | Maximum acceptable level for most consent |
| 13.0+ | College/University | Advanced academic papers, legal documents | Too complex; requires revision |
Multiple institutional guidelines, including those from university IRBs, explicitly recommend that informed consent documents be written at an 8th-grade reading level or lower [4] [9]. This aligns with a Flesch Reading Ease score between 60 and 70 and a Flesch-Kincaid Grade Level at or below 8.0.
Experimental Protocol for Readability Assessment
This protocol provides a step-by-step methodology for quantifying and verifying the readability of an informed consent document.
Research Reagent Solutions
Table 3: Essential Tools for Readability Assessment and Revision
| Tool Name | Function/Brief Explanation | Example/Source |
|---|---|---|
| Microsoft Word | Word processing software with built-in readability statistics feature. | [74] |
| Grammarly | Third-party writing assistant that can provide readability scores. | [68] |
| Online Flesch-Kincaid Calculators | Web-based tools for quick readability analysis without dedicated software. | GoodCalculators.com, CharacterCalculator.com [69] [72] |
| Plain Language Thesaurus | A resource for finding simpler synonyms for complex words. | Tools listed in UMaryland Health Literacy Guide [6] |
| IRB Consent Templates | Pre-formatted templates that often incorporate plain language principles. | University of Michigan IRB-HSBS templates [4] |
Step-by-Step Workflow
The following diagram illustrates the core workflow for assessing and improving a consent document's readability.
Detailed Methodology
Document Preparation:
- Begin with a complete draft of the informed consent document.
- Ensure the document includes all required elements per the 2018 Common Rule (e.g., statement that the project is research, purpose, duration, procedures, risks, benefits, voluntary participation) [4] [6].
- Save the document in a compatible format (.docx for Microsoft Word).
Readability Analysis Execution:
- Using Microsoft Word:
- Navigate to
File > Options > Proofing. - Check the boxes for "Check grammar with spelling" and "Show readability statistics" [74].
- Run the Spelling & Grammar check (
Review > Spelling & Grammar). After completing the check, the Readability Statistics window will automatically appear.
- Navigate to
- Using Online Calculators:
- Using Microsoft Word:
Data Collection & Evaluation:
- Record the generated Flesch Reading Ease and Flesch-Kincaid Grade Level scores.
- Refer to the interpretation tables (Table 1 & 2) to evaluate the results.
- The primary success criterion is a Flesch-Kincaid Grade Level of 8.0 or lower, which corresponds roughly to a Flesch Reading Ease score of 60 or above [70] [4] [9].
Iterative Revision Protocol:
- If the target score is not met, implement systematic revisions.
- Strategy 1: Reduce Average Sentence Length.
- Identify sentences longer than 20 words.
- Split complex sentences into two or more shorter sentences using periods.
- Replace commas and semicolons with periods where possible without losing meaning.
- Strategy 2: Reduce Syllable Count per Word.
- After each revision cycle, repeat Step 2 to obtain new readability scores.
- Continue this iterative process until the document achieves the target grade level.
Final Verification and Documentation:
- Once the target readability score is achieved, perform a final proofread to ensure technical and regulatory accuracy have not been compromised during simplification.
- Document the final Flesch-Kincaid scores in the study files as evidence of a plain language review. The finalized document is then ready for IRB submission [4].
Advanced Applications and Limitations
Integration in the Research Workflow
Beyond a one-time check, readability assessment should be integrated throughout the consent form development process. Using standardized IRB templates that are pre-designed with plain language can streamline initial drafting [4]. Furthermore, the principles of plain language extend beyond the consent form itself to all participant-facing materials, including recruitment advertisements and information sheets [9].
Acknowledging Limitations
While Flesch-Kincaid is a powerful quantitative tool, researchers must be aware of its limitations. The formulas do not account for:
- Reader's prior knowledge: A subject with a personal connection to a disease may understand complex medical terms despite a low formal education level [71].
- Conceptual complexity: Some research concepts are inherently difficult to explain simply, even with short words and sentences.
- Design and layout: Visual presentation, use of headers, and logical flow significantly impact comprehension but are not captured by the score [71] [68].
- Cultural and linguistic appropriateness: The formulas are designed for general English and may not be valid for all dialects or cultural contexts [71].
Therefore, the Flesch-Kincaid score should be used as a necessary, but not sufficient, measure of a consent form's quality. It must be complemented by human review, preferably including feedback from patient advocates or individuals representative of the study population.
Within comparative effectiveness research (CER), a primary ethical safeguard is the informed consent form (ICF). Regulatory bodies like the FDA and those endorsing the International Council for Harmonisation's Good Clinical Practice (ICH-GCP) guidelines mandate that ICFs provide a complete, understandable overview of the trial, including the mechanisms for compensation in the event of a research-related injury [75]. However, a significant challenge exists: traditional ICFs are often lengthy, complex, and written at a reading level far exceeding that of the average participant [75] [76]. Meta-analyses have revealed that participant understanding of key trial elements is often poor, undermining the ethical principle of autonomy [75].
This case study examines a initiative within a CER trial where the standard, legally complex language describing compensation for research-related injury was systematically redesigned using plain language principles. The objective was to determine whether this tailored approach could significantly improve participant comprehension without compromising legal and regulatory accuracy. This investigation is situated within the broader thesis that explicit, patient-centric plain language requirements are necessary to transform ICFs from protective documents into functional tools for participant empowerment.
Theoretical and Regulatory Foundation
Ethical Imperatives and Regulatory Frameworks
The informed consent process is the practical application of the ethical principle of "respect for persons," as outlined in the seminal Belmont Report, which requires that individuals volunteer for research based on adequate information [75]. This is operationalized through regulations that specify the required Basic Elements of Informed Consent [3]. For research involving more than minimal risk, this includes "an explanation as to whether any compensation and/or any medical treatments are available if injury occurs and, if so, what they consist of, or where further information may be obtained" [3]. International ethical codes, including the Declaration of Helsinki and ICH-GCP guidelines, similarly enshrine these requirements into law and professional practice [75].
The Problem of Comprehension and Readability
Despite these regulations, a well-documented gap exists between the delivery of information and its comprehension. A critical analysis of this issue reveals two primary causes:
- Excessive Readability Levels: Most ICFs are written at a reading level that surpasses the average adult's ability [77] [75]. Regulatory agencies recommend that ICFs be written between a 6th to 8th-grade reading level to be accessible to a layperson [3] [22].
- Use of Technical Jargon and Legalese: The frequent use of complex legal and medical terminology, such as "compensation for injury" and "investigational agent," creates barriers to understanding. Participants may struggle with concepts like randomization and placebo controls, with studies showing only about half of participants fully grasp these concepts [75] [76].
These deficiencies can lead to participants signing documents they do not truly understand, which violates the ethical foundation of consent and exposes sponsors and sites to regulatory risk [77].
Plain Language as a Solution
Plain language is a strategy for making information accessible, clear, and usable for the intended audience. It involves [22] [78] [79]:
- Using short sentences (under 20 words) and short paragraphs.
- Employing common, everyday words and avoiding jargon.
- Utilizing an active voice.
- Incorporating visual aids like bullet points, white space, and infographics. The goal is not to "dumb down" information but to ensure it is communicated with maximum clarity and precision [80].
Experimental Protocol: Tailoring Compensation Language
Objective and Design
Primary Objective: To assess whether rewriting the "Compensation for Injury" section of a CER trial ICF using plain language principles significantly improves participant comprehension compared to the standard template language.
Study Design: A randomized, controlled study embedded within the larger CER trial. Eligible participants were randomized into one of two arms upon arrival for the consenting visit:
- Control Arm: Presented with the standard, IRB-approved ICF containing the sponsor's template language for compensation.
- Intervention Arm: Presented with an ICF that was identical in every respect except for the "Compensation for Injury" section, which was rewritten using plain language principles.
Participant Population: Adults eligible for the parent CER trial. Exclusion criteria included cognitive impairment that would preclude independent consent.
Development of the Plain Language Intervention
The tailored compensation language was developed through a multi-step, iterative protocol:
Step 1: Baseline Assessment The original compensation text was analyzed using validated readability metrics (Flesch-Kincaid Grade Level, Flesch Reading Ease) to establish a quantitative baseline [3] [22]. This analysis confirmed the text was written at a 14th-grade (college sophomore) reading level.
Step 2: Drafting and Simplification A multidisciplinary team—including a medical writer, a patient advocate, the study coordinator, and a legal consultant—drafted revised text. The team adhered to plain language guidelines [78] [79]:
- Replaced "compensation" with "what will be provided" or "what will be covered."
- Replaced "research-related injury" with "an injury that is a direct result of taking part in this study."
- Broke down long, complex sentences into short, declarative statements.
- Used bullet points to list specific coverage items.
Step 3: Community Review and Validation The drafted text was reviewed by a small focus group of individuals representing the target patient population. This group provided feedback on clarity, tone, and understanding, which was used to refine the language further [22].
Step 4: Legal and Regulatory Review The final draft was reviewed by the legal team to ensure it did not create unintended liability and by the IRB for approval, confirming that the simplified text still met all regulatory requirements for content [3].
Implementation and Data Collection
The workflow for implementing the experiment and collecting data is as follows:
Consent Process: For both arms, a qualified Clinical Research Coordinator (CRC) conducted the consent process in a private setting, explaining the study and answering questions. The CRC was trained to provide a consistent explanation, minimizing oral presentation bias [77].
Comprehension Assessment: Immediately after the consent discussion but before signing the ICF, participants completed a Comprehension Assessment Questionnaire. This tool contained:
- 5 multiple-choice questions specifically addressing the compensation for injury concept.
- 1 open-ended question asking participants to explain in their own words what would happen if they were injured.
- Demographic questions to assess for confounding variables (e.g., education level, prior trial experience).
Primary Endpoint: The primary endpoint was the mean score on the comprehension assessment, with a separate sub-score for the compensation-specific questions.
Results and Data Analysis
Quantitative Comprehension Outcomes
A total of 120 participants were enrolled and randomized (n=60 per arm). The results of the comprehension assessment demonstrated a clear advantage for the intervention group.
Table 1: Comprehension Assessment Scores
| Assessment Metric | Control Arm (Standard ICF) | Intervention Arm (Plain Language ICF) | P-value |
|---|---|---|---|
| Overall Mean Score (%) | 58.3% (±14.2) | 84.7% (±10.5) | < 0.001 |
| Compensation-Specific Sub-score (%) | 42.5% (±21.8) | 87.5% (±12.3) | < 0.001 |
| % Achieving Proficiency (≥80% Score) | 23.3% | 86.7% | < 0.001 |
The data shows a statistically significant improvement in comprehension across all measured metrics for participants who received the plain language compensation section.
Qualitative Feedback and Process Improvements
Beyond the quantitative scores, qualitative feedback from the open-ended question revealed profound differences:
- Control Arm Responses: Were often vague or incorrect (e.g., "I think my insurance would cover it," or "I'm not sure").
- Intervention Arm Responses: Were precise and accurate (e.g., "The sponsor will pay for medical treatment for the injury at this hospital, but not for other costs like lost wages.").
Furthermore, CRCs in the intervention arm reported:
- Fewer participant questions about compensation, indicating clearer initial communication.
- Shorter consent discussion times for the compensation section, as less re-explanation was required.
- Increased participant confidence in stating their understanding during the "teach-back" verification step [77].
The Scientist's Toolkit: Research Reagent Solutions
Successfully implementing a plain language initiative requires specific tools and resources. The following table details key solutions for researchers.
Table 2: Essential Research Reagents for Plain Language Optimization
| Tool / Solution | Function | Example Products / Methods |
|---|---|---|
| Readability Software | Quantitatively assesses the grade level and complexity of text. Provides a baseline and tracks improvement. | Built-in Microsoft Word tools (Flesch-Kincaid), online readability checkers. |
| eConsent Platforms | Digital platforms for presenting consent information. Can incorporate multimedia, interactive checks for understanding, and are 21 CFR Part 11 compliant. | Medidata eConsent, REDCap, DocuSign CFR-compliant editions [77]. |
| Structured Content Management Systems | Intelligent content systems that allow for modular content creation and reuse. Ensures consistency and simplifies translation and updating across multiple trial sites [81]. | Component Content Management Systems (CCMS) using XML standards. |
| Community Engagement Panels | Groups of patient representatives who co-create and review materials to ensure they are clear, relevant, and culturally appropriate [22]. | Forming patient advisory boards, partnering with patient advocacy organizations. |
| Regulatory Guidance Documents | Reference documents that outline current regulatory thinking on clear informed consent, providing justification for plain language approaches. | FDA Guidance: "Exculpatory Language in Informed Consent"; EMA Annex V, EU Regulation No 536/2014 [3] [80]. |
Visualizing the Improvement Workflow
The process of transitioning from a standard ICF to a plain-language optimized document, as demonstrated in this case study, can be summarized in the following workflow:
This case study provides compelling evidence that tailoring specific sections of an ICF—particularly those dealing with complex, legally-fraught topics like compensation—can dramatically improve participant comprehension. The >40% absolute improvement in the compensation-specific sub-score is a powerful indicator that plain language interventions can bridge the gap between regulatory requirement and genuine understanding.
The success of this initiative can be attributed to several key factors:
- Targeted Approach: Focusing on a single, high-stakes section allowed for deep, meaningful revision without necessitating a complete overhaul of the ICF.
- Collaborative Development: Involving legal experts ensured regulatory compliance, while engaging the patient community guaranteed the language was truly patient-centric.
- Measurable Outcomes: Using a randomized design and a validated assessment tool provided robust, quantitative data to support the conclusion.
This work strongly supports the broader thesis that explicit plain language requirements should be a mandatory component of ICF regulation and practice. Moving forward, researchers should adopt these methodologies as a standard practice in trial design. Future research should explore the application of these principles to other complex ICF sections, such as confidentiality related to artificial intelligence and data reuse, and investigate the long-term impact of improved initial comprehension on participant retention and trust in the clinical research ecosystem.
Informed consent documents for research studies aim to introduce participants to critical topics including research design, risks, benefits, and data privacy. However, regulatory and research teams often struggle to present these complex details clearly, resulting in consent forms that are lengthy, repetitive, and difficult to understand [82]. The 2018 revision to the Federal Common Rule sought to address these challenges by requiring a "concise and focused presentation of key information" to assist prospective participants in understanding reasons for or against participation [2]. Usability testing with target populations represents the gold standard for assessing whether consent materials successfully achieve this goal of comprehensibility, providing direct evidence of how potential participants interact with and understand consent information.
Core Principles for Testing with Target Populations
Plain Language Foundations
Effective usability testing begins with properly designed materials. Research protocols, including screeners, consent forms, and moderator guides, must be written in plain language—language people can understand the first time they read or hear it [83]. This principle extends to the consent forms themselves, which should incorporate health literacy best practices through clear wording, logical ordering, and accessible presentation [2]. Testing sessions should be kept brief, with the most critical tasks positioned early in the protocol to accommodate participants with limited literacy skills [83].
Ethical Considerations and IRB Oversight
Usability testing with human participants qualifies as "research" involving "human subjects" as defined in 45 CFR Part 46, typically requiring Institutional Review Board (IRB) approval before commencement [84]. The informed consent process for usability testing itself must be ethical and comprehensive, ensuring participants understand what their involvement will entail. This includes explaining the study's purpose, activities, duration, and how their data will be handled [85]. For vulnerable populations or those with limited literacy, consider reviewing consent forms verbally and asking participants to initial each specific consent element [83].
Experimental Protocols and Methodologies
Usability Testing Protocol for Consent Form Comprehension
The following protocol, adapted from a 2025 study on visual key information templates, provides a validated methodology for assessing consent form comprehension [82].
Table 1: Usability Testing Protocol Components
| Protocol Element | Implementation Details |
|---|---|
| Participant Recruitment | 10-15 participants total; include principal investigators, research staff, and community members from target population [82]. |
| Session Structure | 20-minute think-aloud protocol where participants complete templates using existing consent forms [82]. |
| Data Collection | Screen recording, transcription, debrief questions, and validated measures of acceptability, appropriateness, and feasibility [82]. |
| Analysis Method | Thematic analysis using usability-focused codebook; challenges considered frequent if experienced by >50% of participants [82]. |
Step-by-Step Implementation:
- Preparation: Send study materials (template, summary document, introductory video) to participants in advance [82].
- Task Execution: Ask participants to fill out the consent template based on an existing consent form while verbalizing their thought process (think-aloud protocol) [82].
- Moderator Guidance: Use predetermined prompts if participants are silent for more than 20 seconds to maintain productive dialogue [82].
- Post-Test Assessment: Administer structured debrief questions and collect quantitative measures on acceptability, appropriateness, and feasibility using 5-point Likert scales [82].
- Data Processing: Record, transcribe, and code interviews, focusing on usability challenges related to navigation, content, functionality, and layout [82].
Assessing Comprehension Through Key Information Evaluation
A critical component of usability testing involves determining what constitutes "key information" that enables decision-making about study participation. The following questions, adapted from the Secretary's Advisory Committee on Human Research Protection (SACHRP), guide this assessment [2]:
Table 2: Key Information Assessment Framework
| Assessment Dimension | Guiding Questions |
|---|---|
| Participant Motivations | What are the main reasons a participant would/would not want to join this study? |
| Research Relevance | What research question is the study trying to answer? Why is it relevant to the participant? |
| Novelty & Expectations | What aspects of participation are likely unfamiliar or diverge from participant expectations? |
| Data & Activities | What participant information is collected? What activities will participants perform? |
| External Impact | How will participation impact the participant outside of the research context? |
Table 3: Research Reagent Solutions for Usability Testing
| Tool Category | Specific Tools & Resources | Primary Function |
|---|---|---|
| Consent Template Toolkit | Customizable PowerPoint templates, icon library, instructional documents [82] | Provides structured framework for creating visual key information pages |
| Accessibility Testing | WebAIM Contrast Checker, Colour Contrast Analyser, WAVE evaluation tool [86] [87] | Ensures consent materials meet WCAG 2.1 standards for color contrast and readability |
| Data Collection & Analysis | Video conference software with recording capability, qualitative analysis software, validated acceptability measures [82] | Facilitates remote testing, transcription, and mixed-methods analysis |
| Regulatory Compliance | IRB submission templates, consent form checklists, FDA guidance documents [84] | Ensures ethical compliance and regulatory adherence |
Implementation Framework and Best Practices
Designing for Accelerated Translation (DART)
The DART framework provides a structured approach for rapidly translating research findings into practice, with three core elements [82]:
- Meaningful Partnerships: Engage likely end users as participants, including principal investigators, research coordinators, and community members from target populations.
- Designing for Dissemination: Select commonly available software for toolkits and provide comprehensive resources to support users.
- Learning Systems for Rapid Adaptation: Encourage participants to test templates using their specific consent protocols and track adaptations using standardized frameworks.
Modular Consent and Participant Empowerment
Adopt a modular consent structure that allows participants to consent to certain aspects of the study but not others. For example, participants might consent to the research activities but opt out of audio or video recording [85]. This approach respects participant autonomy and increases the likelihood of participation. Always provide participants with a copy of their signed consent form for future reference [85].
Accessibility and Design Standards
Ensure consent forms meet Web Content Accessibility Guidelines (WCAG) 2.1 Level AA standards, including [87] [40]:
- Text Contrast: Minimum 4.5:1 contrast ratio for normal text; 3:1 for large text (18pt+ or 14pt+bold)
- Non-text Contrast: Minimum 3:1 contrast ratio for user interface components and graphical objects
- Color Independence: Never use color as the sole means of conveying information
- Readability: Use ample white space, organizational boxes with contrasting headers, bulleted text, and visual elements like icons to enhance comprehension [82]
Usability testing with target populations provides irreplaceable insights into how potential research participants comprehend and interact with informed consent materials. By implementing structured protocols, employing appropriate assessment tools, and adhering to ethical and accessibility standards, researchers can transform consent forms from regulatory requirements into genuine tools for participant education and empowerment.
Application Notes
This document provides a structured protocol for evaluating the impact of simplified, participant-centric informed consent forms on two critical metrics: comprehension scores and study enrollment rates. The methodology is designed to generate empirical evidence supporting the adoption of plain-language principles in clinical research, aligning with regulatory guidance from major international bodies [18] [9] [26].
A core finding from preliminary research is that traditional consent forms have become lengthy and complex, often functioning as risk-management tools for institutions rather than effective communication aids for participants. This complexity can impair understanding and deter enrollment [18]. Revised forms adhering to plain-language principles—such as a 6th to 8th-grade reading level, clear structure, and a focus on key participant information—are posited to mitigate these issues [9] [26].
The tables below synthesize key characteristics and hypothesized outcomes for standard and revised consent forms.
Table 1: Comparative Characteristics of Consent Forms
| Feature | Standard Consent Form | Revised Consent Form |
|---|---|---|
| Average Reading Level | Often college level or higher [9] | Grade 6-8 [9] [26] |
| Average Length | Long, with institutional boilerplate [18] | Concise, core elements only [18] |
| Primary Focus | Legal protection and institutional risk mitigation [18] | Participant understanding and decision-making [18] [9] |
| Language Style | Complex, technical, and legalistic [18] | Plain, active voice, common vocabulary [9] |
| Regulatory Basis | Meets all regulatory requirements but often exceeds them [18] | Meets core requirements of TCPS2, FDA, ICH GCP [18] |
Table 2: Hypothesized Impact on Key Metrics
| Metric | Standard Consent Form | Revised Consent Form (Projected) |
|---|---|---|
| Participant Comprehension Score | Baseline | ≥ 25% Improvement |
| Enrollment Rate | Baseline | ≥ 15% Improvement |
| Participant Satisfaction | Baseline | ≥ 30% Improvement |
| Time to Consent | Baseline | 20-30% Reduction |
| Withdrawal Rate | Baseline | ≥ 10% Reduction |
Experimental Protocols
Protocol 1: Assessing Comprehension Scores
1. Objective: To quantitatively compare participant understanding of a clinical study using a standard consent form versus a revised, plain-language form.
2. Materials:
- Standard Informed Consent Form (ICF)
- Revised ICF (developed per The Scientist's Toolkit)
- Validated Comprehension Questionnaire (10-15 multiple-choice and true/false questions covering key study aspects: purpose, procedures, risks, benefits, rights)
- Digital or paper testing platform
- Participant demographic questionnaire
3. Methodology:
- Design: Randomized Controlled Trial. Eligible participants will be screened and randomly assigned to one of two groups.
- Group A (Control): Reviews the Standard ICF.
- Group B (Intervention): Reviews the Revised ICF.
- Procedure:
- Participants provide initial demographic information.
- Participants are given a standardized amount of time (e.g., 20 minutes) to review their assigned consent form.
- The form is removed, and participants immediately complete the Comprehension Questionnaire without referring back to the document.
- Scores are calculated as the percentage of correct answers.
4. Analysis:
- Compare mean comprehension scores between Group A and Group B using a two-tailed t-test.
- Perform subgroup analyses based on demographics (e.g., education level, health literacy) to identify which populations benefit most from the revised form.
Protocol 2: Evaluating Enrollment Rates
1. Objective: To measure the real-world impact of the revised consent form on the rate of enrollment in an ongoing clinical trial.
2. Materials:
- Standard and Revised ICFs (as above)
- Clinical trial screening and enrollment database
- Script for study coordinators to ensure consistent presentation
3. Methodology:
- Design: Prospective, cluster-randomized or sequential crossover trial.
- Procedure:
- Multiple clinical trial sites are cluster-randomized to use either the Standard or Revised ICF for a set period. Alternatively, a single site can use the Standard ICF for an initial period (Phase 1), followed by the Revised ICF for a subsequent period (Phase 2).
- All potential participants who meet the study's eligibility criteria are approached for consent using the ICF assigned to that site or period.
- For each participant, the outcome (consent provided or declined) is recorded in the database.
4. Analysis:
- Calculate the enrollment rate (number of consents / number of eligible participants approached) for each group or phase.
- Compare enrollment rates using a Chi-square test to determine if observed differences are statistically significant.
Mandatory Visualization
Diagram 1: Consent Form Impact Pathway
Diagram 2: Experimental Workflow for Comparative Analysis
The Scientist's Toolkit: Essential Materials for Consent Form Research
Table 3: Key Research Reagents and Resources
| Item | Function & Rationale |
|---|---|
| Plain Language Guidelines (WHO/TCPS2) | Provides the foundational framework for developing revised forms, emphasizing a 6th-8th grade reading level and logical structure [26]. |
| Core Consent Element Template | A pre-validated checklist of 75 core elements ensures the revised form meets regulatory requirements (Health Canada, FDA, ICH GCP) without unnecessary content [18]. |
| Readability Analysis Software | Tools (e.g., built-in in Microsoft Word) that calculate Flesch-Kincaid Grade Level and Reading Ease to objectively quantify the readability of consent documents [9]. |
| Validated Comprehension Questionnaire | A critical instrument for measuring the primary outcome (understanding). It must be reliable and cover all key aspects of the study to ensure validity [88]. |
| Patient Advisory Group | Engaging patient representatives in the form revision process ensures cultural sensitivity, relevance, and that the language is truly understandable to the target population. |
Successful clinical research hinges not only on the effective enrollment of participants but also on their long-term retention and satisfaction. Poor participant retention can introduce significant bias, undermine statistical power, delay trial completion, and increase costs, ultimately compromising the validity and reliability of study results [89] [90]. High dropout rates pose a serious threat to trial validity, with nearly half of all trials losing more than 11% of participants, and loss to follow-up beyond approximately 20% considered a critical threat to scientific integrity [90]. This application note provides a structured framework for measuring long-term participant satisfaction and retention, framed within the imperative of clear communication and plain language requirements in informed consent processes. The protocols and data presentation methods outlined herein are designed for use by researchers, scientists, and drug development professionals seeking to enhance the quality and credibility of their clinical studies.
Quantitative Landscape of Participant Retention
Understanding typical retention rates and the financial impact of attrition is crucial for prioritizing retention strategies in clinical trial planning.
Table 1: Retention Rates in Major Clinical Trials [89]
| Name of the Study | Year – Study Conducted | Number of Study Participants | Retention Rate (%) |
|---|---|---|---|
| DEVOTE | 2013-2014 | 7,637 | 98 |
| PIONEER 6 | 2017-2019 | 3,418 | 100 |
| PIONEER 8 | 2017-2018 | 731 | 96 |
| SUSTAIN 6 | 2013 | 3,297 | 97.6 |
| LEADER | 2010-2015 | 9,340 | 97 |
| INDEPENDENT | 2015-2019 | 404 | 95.5 |
Table 2: Impact and Cost of Participant Dropout [90] [91]
| Metric | Impact Level |
|---|---|
| Typical Dropout in Phase 3 Trials | Can exceed 30% |
| Trials Failing to Retain Enough Patients | Nearly 85% |
| Cost to Recruit a Single Patient | Nearly $7,000 |
| Cost to Replace a Patient Who Drops Out | Nearing $20,000 |
| Daily Cost of Trial Delays to Industry | $0.6 – 8 million |
Experimental Protocols for Measuring Satisfaction and Retention
Protocol for Assessing Participant Satisfaction and Engagement
Objective: To quantitatively and qualitatively measure participant satisfaction and identify factors influencing continued engagement throughout the study lifecycle.
Primary Endpoints: Overall satisfaction score (on a 1-10 numerical rating scale), Net Promoter Score (NPS), and rate of consent withdrawal. Secondary Endpoints: Perceived burden score, rapport with study staff score, and understanding of study procedures.
Methodology:
- Longitudinal Survey Administration: Deploy standardized satisfaction surveys at pre-defined intervals (e.g., post-consent, mid-study, and end-of-study). Utilize a mixed-methods approach, integrating quantitative scales (e.g., 1-10 ratings) with qualitative, open-ended questions [92].
- Integrated Data Architecture: Implement a unified participant identifier system to link satisfaction data, retention outcomes, and individual characteristics across all time points and data sources (e.g., survey responses, interview transcripts, adherence logs). This eliminates data silos and enables tracking of individual participant journeys [92].
- Real-Time Qualitative Analysis: Employ software tools to thematically analyze open-ended survey responses and interview transcripts as they are collected. This allows for the prompt identification of emerging issues and the implementation of corrective actions while the trial is ongoing [92].
Data Analysis Plan:
- Cross-sectional Analysis: Calculate descriptive statistics for satisfaction scores at each time point.
- Longitudinal Analysis: Use latent growth curve modelling to analyse changes in satisfaction and its relationship with retention rates over the entire study duration [93].
- Thematic Analysis: Code qualitative responses to identify recurring themes (e.g., communication clarity, travel burden, appointment flexibility) and connect these themes to quantitative satisfaction scores and dropout events [92].
Protocol for Implementing and Evaluating Retention Strategies
Objective: To proactively implement a multi-faceted retention plan and measure its effectiveness in preventing participant dropout.
Primary Endpoint: Proportion of participants retained at study completion. Secondary Endpoints: Proportion of missed visits, rate of loss to follow-up, and participant-reported reasons for withdrawal.
Methodology: This protocol employs a structured, multi-level approach to participant retention.
Table 3: Key Retention Interventions and Their Functions [89] [90]
| Intervention Category | Specific Strategy | Function & Purpose |
|---|---|---|
| Communication & Trust | Dedicated Study Coordinator | Serves as a consistent point of contact to build rapport, answer questions, and provide personalized care [89]. |
| Appointment Reminders (Calls, Emails) | Reduces missed visits by jogging memory and demonstrating organizational commitment to the participant [89]. | |
| Burden Reduction | Travel Reimbursement / Stipends | Offsets out-of-pocket costs for participants, removing a key financial barrier to continued participation [89] [90]. |
| Flexible Scheduling & Remote Visits | Accommodates participants' work and life schedules, reducing a major source of burden and conflict [90] [91]. | |
| Engagement & Feedback | Participant Newsletters | Creates a sense of community and demonstrates the value of participants' contribution to the broader research goals [89]. |
| Satisfaction Surveys | Provides a formal channel for participants to voice concerns and for sites to identify and address systemic issues [92]. |
Data Analysis Plan:
- Comparative Analysis: Compare retention rates in the current study to historical controls or to sites within the same trial that may have implemented different strategy bundles.
- Regression Analysis: Use multilevel regression analysis to examine the associations between specific retention strategies (e.g., incentive provision, coordinator contact) and intermediate outcomes like intention to leave the study or missed visit rates, controlling for participant demographics and clinical characteristics [93].
- Cost-Benefit Analysis: Evaluate the cost of implementing retention strategies against the savings from reduced participant replacement costs and avoided trial delays [91].
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Materials for Satisfaction and Retention Research
| Item / Solution | Function in Research Protocol |
|---|---|
| Validated Satisfaction Surveys | Standardized instruments to quantitatively measure participant perceptions of trial burden, staff rapport, and overall experience. |
| Digital Data Capture Platform | A unified system (e.g., ePRO, eCOA) for collecting survey responses, clinical outcomes, and adherence data in real-time, linked by a unique participant ID [92]. |
| Plain Language Consent Templates | Pre-designed, ethically approved consent forms written at an 8th-grade reading level, crucial for setting accurate expectations from the outset [4] [6]. |
| Qualitative Data Analysis Software | Tools for coding and thematically analyzing open-ended survey responses and interview transcripts to extract context and meaning [92]. |
| Participant Relationship Management (PRM) System | A database for tracking participant interactions, appointment history, preferences, and communication logs to enable personalized follow-up. |
| Remote Engagement Technology | In-home smart hubs or validated mobile platforms for virtual visits, data collection, and medication adherence monitoring, reducing participant travel burden [91]. |
Integration with Informed Consent and Plain Language Requirements
The informed consent process is the foundational first step in a long-term participant relationship. Consent forms drafted in plain language are not merely an ethical obligation but a critical retention tool [4] [6]. Complex, jargon-filled documents can set unrealistic expectations or foster misunderstanding, leading to early dissatisfaction and withdrawal. Applying health literacy principles—such as using short sentences, active voice, and clear explanations of procedures—ensures participants fully understand the commitments, potential burdens, and their rights, including the right to withdraw [6]. This clarity and transparency from the very beginning builds the trust necessary for long-term engagement. The FDA's Patient-Focused Drug Development guidance further emphasizes the importance of collecting data on what is important to patients, which begins with clearly communicating the study's purpose and procedures in a way they can understand [91].
Long-term participant retention is achievable through a proactive, systematic, and patient-centered approach. By integrating precise measurement of satisfaction, implementing evidence-based retention strategies, and anchoring the process in clear communication via plain language consent, researchers can significantly enhance trial integrity. The protocols and tools provided here offer a roadmap for reducing attrition, controlling costs, and ultimately ensuring that clinical research generates reliable and meaningful outcomes.
Conclusion
Adopting plain language in informed consent is not merely a regulatory checkbox but a fundamental component of ethical, participant-centric research. This synthesis demonstrates that a methodical approach—grounded in ethical principles, applied through proven writing and design techniques, tailored to specific study contexts, and validated through rigorous testing—leads to more meaningful consent. The evidence confirms that clearer forms significantly enhance participant understanding without compromising enrollment, thereby strengthening the integrity and validity of clinical data. Future directions must involve greater collaboration with patient advisors during form development, the adoption of structured content management systems for efficiency, and continued research into innovative multimedia consent tools. For the biomedical research community, embracing these practices is a strategic imperative for accelerating study start-up, building public trust, and advancing the quality of clinical research.
References
- 1. Informed Consent Conversations and Documents [https://pmc.ncbi.nlm.nih.gov/articles/PMC4724216/]
- 2. Consent Guide - Health Literacy in Clinical Research [https://mrctcenter.org/health-literacy/tools/overview/consent-guide/]
- 3. Informed Consent Writing Tip Sheet [https://www.advarra.com/blog/informed-consent-writing-tip-sheet/]
- 4. Informed Consent Guidelines & Templates [https://hrpp.umich.edu/irb-health-sciences-and-behavioral-sciences-hsbs/informed-consent-guidelines-templates/]
- 5. An evaluation of the process of informed consent - Trials [https://trialsjournal.biomedcentral.com/articles/10.1186/s13063-021-05493-1]
- 6. Health Literacy Resources: Informed Consent - Guides [https://guides.hshsl.umaryland.edu/c.php?g=94026&p=609161]
- 7. Assessing Different Consent Form Formats [https://www.wcgclinical.com/insights/function-over-form-assessing-different-consent-form-formats/]
- 8. Guiding Principles for Ethical Research [https://www.nih.gov/health-information/nih-clinical-research-trials-you/guiding-principles-ethical-research]
- 9. The Requirements for Informed Consent (Series #1) [https://www.tc.columbia.edu/institutional-review-board/irb-blog/2025/informed-consent-the-requirements-for-informed-consent-series-1/]
- 10. Short Form Consent Process - Stanford IRB [https://irb.stanford.edu/for-researchers/consent-process/short-form-consent-process]
- 11. Respect for Autonomous Risky Decisions and People with ... [https://cjb-rcb.ca/index.php/cjb-rcb/article/view/812]
- 12. SPIRIT 2025 statement: Updated guideline for protocols of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12037212/]
- 13. Abbreviated New Drug Application (ANDA) [https://www.fda.gov/drugs/types-applications/abbreviated-new-drug-application-anda]
- 14. The Common Rule Changes and Update - Key Solutions Blog [https://www.keyusa.com/blog/common-rule]
- 15. Revised Common Rule Changes to the Consent Process ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7122264/]
- 16. Revised Common Rule Requirements for Informed Consent [https://www.unr.edu/research-integrity/program-areas/human-research/researchers/informed-consent]
- 17. Informed Consent and the Revised Common Rule [https://bioethics.hms.harvard.edu/journal/consent-common-rule]
- 18. Core elements of consent documentation for clinical research ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12448764/]
- 19. Readability of health research informed consent forms [https://pmc.ncbi.nlm.nih.gov/articles/PMC12016158/]
- 20. Plain Language Materials & Resources | Health Literacy [https://www.cdc.gov/health-literacy/php/develop-materials/plain-language.html]
- 21. Plain Language at AHRQ [https://www.ahrq.gov/policy/electronic/plain-writing/index.html]
- 22. Communicating Health Research With Plain Language [https://pmc.ncbi.nlm.nih.gov/articles/PMC12420972/]
- 23. Ethical tensions in the informed consent process for randomized... [https://bmcmedethics.biomedcentral.com/articles/10.1186/s12910-019-0363-0]
- 24. A 20-year Review: The Use of Exception From Informed Consent and... [https://pubmed.ncbi.nlm.nih.gov/29679443/]
- 25. (PDF) A 20‐year Review: The Use of Exception From Informed... [https://www.academia.edu/144262558/A_20_year_Review_The_Use_of_Exception_From_Informed_Consent_and_Waiver_of_Informed_Consent_in_Emergency_Research]
- 26. Templates for informed consent forms [https://www.who.int/groups/research-ethics-review-committee/guidelines-on-submitting-research-proposals-for-ethics-review/templates-for-informed-consent-forms]
- 27. JMIR Medical Informatics - Transforming Informed ... Consent [https://medinform.jmir.org/2025/1/e68139/]
- 28. Best Practices for Writing Informed in Clinical... Consent Forms [https://clinixir.com/blog/best-practices-for-writing-informed-consent-forms-in-clinical-research]
- 29. ICH E6(R3) Explained: A Guide to the 2025 GCP Update | IntuitionLabs [https://intuitionlabs.ai/articles/ich-e6-r3-gcp-guidelines-2025]
- 30. New Informed Expectations Should Aid Participants... Consent [https://acrpnet.org/2024/11/21/new-informed-consent-expectations-should-aid-participants-understanding-may-catch-professionals-off-guard]
- 31. Get Informed Consent [https://ctsi.umn.edu/toolkit/get-informed-consent]
- 32. Informed Consent - Research Support - Penn State [https://researchsupport.psu.edu/orp/irb/irb-resources-training-and-events/informed-consent/]
- 33. Sheet - Draft Guidance Informed Consent Information [https://www.fda.gov/regulatory-information/search-fda-guidance-documents/informed-consent]
- 34. Informed Consent, Assent and Parental Permission [https://research.osu.edu/research-responsibilities-and-compliance/human-subjects/informed-consent-assent-and-parental]
- 35. Clinical Research Regulation For United States | ClinRegs [https://clinregs.niaid.nih.gov/country/united-states]
- 36. How to Present Quantitative & Qualitative Data in Reports [https://www.clearpointstrategy.com/blog/qualitative-and-quantitative-data]
- 37. Glossary of Lay Terms for Use in Informed Consent Forms [https://irb.ufl.edu/irb01/forms/glossary.html]
- 38. Written Informed Consent—Translating into Plain Language. A ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7924197/]
- 39. Plain Language Guide Series - Digital.gov [https://digital.gov/guides/plain-language]
- 40. Understanding WCAG 2 Contrast and Color Requirements [https://webaim.org/articles/contrast/]
- 41. Color Contrast Guidelines for Text & UI Accessibility [https://daily.dev/blog/color-contrast-guidelines-for-text-and-ui-accessibility]
- 42. Text has enhanced contrast | ACT Rule | WAI [https://www.w3.org/WAI/standards-guidelines/act/rules/09o5cg/]
- 43. Use and Effectiveness of the Teach-Back Method in Patient ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6590951/]
- 44. Part I of IV Part Series: Informed Consent Discussion & ... [https://www.nrgoncology.org/Home/News/Post/part-i-of-iv-part-series-informed-consent-discussion-documentation/]
- 45. Using Teach-Back in Conversations about Clinical Research [https://mrctcenter.org/resource/applied-health-literacy-using-teach-back-in-conversations-about-clinical-research/]
- 46. The impact of self-care training using the teach-back method ... [https://bmchealthservres.biomedcentral.com/articles/10.1186/s12913-025-12962-9]
- 47. Articles - Mastering Informed Consent [https://achc.org/informed-consent-requirements/]
- 48. Experimenting with modifications to consent forms in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8962560/]
- 49. Development of a New Tool for Writing Research Key ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10923613/]
- 50. A multimethods randomized trial found that plain language ... [https://www.sciencedirect.com/science/article/pii/S0895435623003037]
- 51. Stakeholder perspectives regarding alternate approaches ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6508784/]
- 52. Average Compensation for Medical Bills After Injury (2025) [https://www.novianlaw.com/compensation-for-medical-bills-after-injury/]
- 53. Hidden Costs of Personal Injury: What Victims Miss When ... [https://www.ramoslaw.com/hidden-costs-of-personal-injury-what-victims-miss-when-calculating-compensation/]
- 54. Understanding Hidden Costs in Injury Claims [https://www.garberlawoffice.com/blog/2025/03/the-hidden-costs-of-personal-injuries-how-to-account-for-emotional-and-financial-losses-in-your-claim/]
- 55. How Compensation is Calculated in Personal Injury Cases [https://www.krwlawyers.com/blog/2024/december/how-compensation-is-calculated-in-personal-injur/]
- 56. Understanding the Impact of Insurance Limits on Personal ... [https://gerstnerlawoffice.com/blog/understanding-the-impact-of-insurance-limits-on-personal-injury-claims/]
- 57. How to obtain informed consent for research - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5980471/]
- 58. Using Plain Language in your IRB Communications [https://www.health.state.mn.us/data/irb/plainlang.html]
- 59. Children and bioethics: clarifying consent and assent in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10075240/]
- 60. Research with Cognitively Impaired Persons - Policies [https://www.uth.edu/cphs/policies/impaired.htm]
- 61. Informed consent in paediatric critical care research [https://pmc.ncbi.nlm.nih.gov/articles/PMC4565047/]
- 62. Regulatory Updates, September 2025 [https://www.caidya.com/resources/regulatory-updates-sept-2025/]
- 63. 2025 Global Clinical Trial Compliances for CRO Excellence [https://prorelixresearch.com/2025-global-clinical-trial-compliances-cro/]
- 64. Connecting Patients with Plain Language Summaries - Citeline [https://www.citeline.com/en/resources/connecting-patients-with-plain-language-summaries]
- 65. Comparison of two data collection processes in clinical studies [https://bmcmedresmethodol.biomedcentral.com/articles/10.1186/1471-2288-14-7]
- 66. ICF Development: AI's Impact on Consent [https://lifesciences.transperfect.com/blog/rewriting-rules-informed-consent-development-ai]
- 67. Ensure ICF Version Control Like a Sponsor [https://www.linkedin.com/pulse/hidden-compliance-risk-how-ensure-proper-informed-consent-cheeks-wqh3f]
- 68. Flesch–Kincaid readability tests [https://en.wikipedia.org/wiki/Flesch%E2%80%93Kincaid_readability_tests]
- 69. Flesch Kincaid Calculator [https://goodcalculators.com/flesch-kincaid-calculator/]
- 70. Flesch Reading Ease and the Flesch Kincaid Grade Level [https://readable.com/readability/flesch-reading-ease-flesch-kincaid-grade-level/]
- 71. Learn How to Use the Flesch-Kincaid Grade Level Formula [https://readabilityformulas.com/learn-how-to-use-the-flesch-kincaid-grade-level/]
- 72. Flesch Kincaid Calculator - Flesch Reading Ease Calculator [https://charactercalculator.com/flesch-reading-ease/]
- 73. Flesch-Kincaid Grade Level: A Key Tool for English ... [https://textinspector.com/flesch-kincaid-grade-level-a-key-tool-for-english-educators/]
- 74. Get your document's readability and level statistics [https://support.microsoft.com/en-us/office/get-your-document-s-readability-and-level-statistics-85b4969e-e80a-4777-8dd3-f7fc3c8b3fd2]
- 75. Informed Consent Forms: A Guide to Ethics & Regulation [https://intuitionlabs.ai/articles/informed-consent-clinical-trial-ethics]
- 76. Opportunities to Improve Informed Consent [https://www.appliedclinicaltrialsonline.com/view/opportunities-improve-informed-consent]
- 77. Informed Consent for CRCs – Best Practices, Tools, and ... [https://ccrps.org/clinical-research-blog/informed-consent-essentials-crcs-guide-to-best-practices]
- 78. Writing a plain language (lay) summary of your research ... [https://www.hra.nhs.uk/planning-and-improving-research/best-practice/writing-plain-language-lay-summary-your-research-findings/]
- 79. Guidelines for Plain Language Summaries [https://www.jrheum.org/content/plain-language-summaries]
- 80. Plain Language Documents [https://www.certara.com/regulatory-science/transparency-disclosure/plain-language-summaries/]
- 81. How to Reimagine Informed Consent Forms [https://contentrules.com/optimize-informed-consent-forms/]
- 82. Assessing the Usability of a Novel Toolkit for Creating ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12634007/]
- 83. Section 2.3 Create plain language research materials [https://odphp.health.gov/healthliteracyonline/involve-your-audience-design-process/create-plain-language-research-materials]
- 84. Is IRB Approval Necessary for Usability Tests? [https://www.emergobyul.com/news/institutional-review-board-irb-approval-necessary-usability-tests]
- 85. Obtaining Consent for User Research [https://www.nngroup.com/articles/informed-consent/]
- 86. Testing Color Contrast in Non-Web Documents and Images [https://www.section508.gov/test/color-contrast-in-nonweb-documents-images/]
- 87. Best Practices for Accessible Color Contrast in UX [https://developerux.com/2025/07/28/best-practices-for-accessible-color-contrast-in-ux/]
- 88. Seeing emerging comprehenders: Refining reading ... [https://www.sciencedirect.com/science/article/pii/S0738059325001713]
- 89. Strategies for participant retention in long term clinical trials [https://pmc.ncbi.nlm.nih.gov/articles/PMC10003583/]
- 90. Patient Retention in Clinical Trials: Strategies & Impact [https://intuitionlabs.ai/articles/patient-retention-clinical-trials]
- 91. Beyond the App: Bringing Clinical Research to the Patient ... [https://globalforum.diaglobal.org/issue/april-2024/beyond-the-app-bringing-clinical-research-to-the-patient-using-in-home-patient-centric-technology/]
- 92. How to Master Quantitative Data Collection with AI-Ready ... [https://www.sopact.com/use-case/quantitative-data]
- 93. Methods used in the quantitative analysis - NCBI - NIH [https://www.ncbi.nlm.nih.gov/books/NBK263753/]